![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
|
Cystic fibrosis in Uruguay
ABSTRACT. We conducted clinical and genetic analyses of 52 cystic fibrosis (CF) patients in Uruguay, which is about half of the known affected individuals in the country. A relatively high proportion had a mild presentation, characterized by pancreatic sufficiency (28%), a strong pulmonary component (97%), and borderline sweat electrolyte measurements (25%). Mutational analysis of CF chromosomes demonstrated a relatively low incidence of the DF508 allele (40%) and a large number of other cystic fibrosis conductance regulator mutations, with an overall detection rate of about 71%. Fifteen different mutations were detected in our patients: DF508, G542X, R1162X, G85E, N1303K, R334W, R75Q, R74W, D1270N, W1282X, DI507, 2789+5G®A, R1066C, -816C/T, R553X, as well as RNA splicing variant IVS8-5T. This group of Uruguayan CF patients has some characteristics in common with other populations of similar origin (Hispanics), as well as some unique characteristics Key words: Cystic fibrosis, Autosomal genetic disease, Cystic fibrosis transmembrane conductance regulator mutations
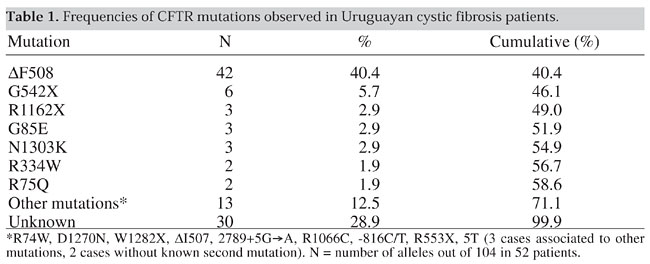
INTRODUCTION Cystic fibrosis (CF) is a recessive, autosomal genetic disease most common among Caucasian populations and caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene (Welsh et al., 1995). The DF508 deletion (frequency of 68% worldwide) is the most common mutation, but the proportion of this allele and other CFTR mutations varies considerably among populations and geographic locations (Tsui, 1992; The Cystic Fibrosis Genetic Analysis Consortium, 1994; Welsh et al., 1995). There is much clinical heterogeneity in CF, which can be partially explained by the various CFTR genotypes (Zielenski and Tsui, 1995). Little has been published on cystic fibrosis in Latin America (Valenzuela, 1988; Raskin et al., 1993; Orozco et al., 1993; Ríos et al., 1994; Chertkoff et al., 1997). It is difficult to compare the known populations to that of Uruguay due to differences in ethnic compositions. Latin Americans are principally a combination of three populations: Amerindians, Africans and Caucasians. The genetic characteristics of South American admixed populations vary throughout the continent according to location, origin of the Caucasian or African immigrants, and the distribution of Amerindians. Uruguay is a small country (130,000 km2) located between Argentina and Brazil. It has about 3,200,000 inhabitants (INE, 1996), composed mainly of Caucasians (86% or more) with 6-11% African-derived individuals and 1-2% Amerindian-derived individuals (Sans et al., 1997). Very low proportions of ethnically non-admixed Amerindians remain, and no isolated groups remained after the middle of the last century (González and Rodríguez, 1992). Africans were introduced into Uruguay as slaves between 1743 and 1842. In 1829, there were an estimated 8,500 African people among approximately 74,000 inhabitants (Sans and Barreto, 1998; Pi and Vidart, 1969). There were several waves of European immigration to Uruguay in different periods. Europeans came from Spain (mainly the Canary Islands, Galicia and Asturias), the Basque region, Italy, Portugal and England. More recently, Central-European Jewish immigrants also came to the country. Uruguay has a small population and a low birth rate (18.9/1000), with a CF prevalence of approximately 1/9600 inhabitants (H. Cardoso, unpublished results). Accordingly, the resulting CF population is relatively small (approximately 100 registered individuals) and it was possible to study a considerable proportion of it. MATERIAL AND METHODS A sample of 104 chromosomes corresponding to 52 unrelated, clinically confirmed CF patients, was selected from a group of 160 individuals. These patients were referred to us for molecular diagnosis or to confirm a clinical diagnosis. Genetic counseling was provided to the family. The patients were diagnosed according to the following criteria: a) Presence of lung disease. Each subject was considered to have pulmonary symptomatology when at least one of the following was observed: bronchiectasis, atelectasis, hyperinflation, airway obstruction, recurrent/chronic pneumonia, Pseudomonas sp. infections or nasal polyps. b) Presence of gastrointestinal and nutritional abnormalities. These were defined by: chronic diarrhea, failure to thrive, meconium ileus and rectal prolapse. In our patients pancreatic insufficiency was determined by abnormal fecal fat values or the need for pancreatic enzyme replacement therapy. c) High sweat chloride concentrations. At least two sweat tests were made on each patient (Gibson and Cooke, 1959). A borderline sweat test value (40-60 mEq/l) or a negative test (<40 mEq/l) did not preclude molecular analysis if pulmonary and/or gastrointestinal symptoms were also present. The CFTR mutations were detected by using one or more of the following methods: a) Reverse hybridization technique for eight mutations frequent in Europe (DF508, G542X, N1303K, 1717-1G®A, W1282X, G551D, R553X and DI507), using a commercial kit from Inno Lipa CF2, Innogenetics, Belgium. b) The G85E, R334W and 3120+1G®A mutations, common in the Spanish population, were analyzed by restriction enzyme analysis (Gasparini et al., 1991; Zielenski et al., 1991). c) Single strand conformation polymorphism (SSCP) according to Orita et al. (1989). d) Multiplex heteroduplex analysis using the mutation detection enhancement gel matrix (mHET; Aznarez et al., 1998) e) Denaturing gradient gel electrophoresis (DGGE; Fanen et al., 1992). In c, d and e, the mutation characterization was performed by direct sequencing of the implicated DNA fragments (Sanger and Coulson, 1975). Patients were tested for the CFTRdele2,3 (21kb) allele (Dork et al., 2000) and variants in the T-tract of the branch/acceptor splice signal of intron 8 (Chillón et al., 1995). RESULTS Genetic analysis led to the detection of 15 different mutations: DF508, G542X, R1162X, G85E, N1303K, R334W, R75Q, R74W, D1270N, W1282X, DI507, 2789+5G®A, R1066C, R553X and -816C/T. In addition to these well-defined CFTR mutations, RNA splicing chromosomes carrying the IVS8-5T variant were also detected. The mutations that had frequencies above 1% are shown in Table 1.
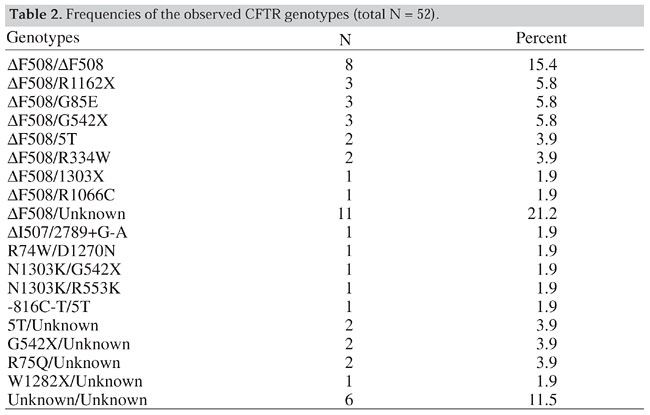
The most prevalent mutation, DF508, was found in 42/104 CF chromosomes, with the second most common allele being the G542X (6/104). Although we used multiple techniques, about 29% of the mutations remained unidentified (Table 1). Besides the listed mutations the following polymorphisms were also present in our patients: -895G-T, 1001+11C/T, 1540A/G, 1898+152T/A, 875+40A/G, 3041-92G/A, 2377C/T and 3212T/C. The different CFTR genotypes are shown in Table 2. The most frequent was DF508/DF508 (8/52). Other common genotypes were: DF508/other, found in 15 patients, DF508/unknown observed in 11, other/other, in 6 and unknown/unknown, in 6. Of the 52 patients tested, 28 had both mutations detected, 18 only one mutation known, and in 6 both mutations unknown. T-tract analysis revealed the IVS8-5T variant pairing with DF508 (2 patients) and -816C/T mutations (one patient), as well as unknown mutations in 2 patients.
All individuals had pulmonary symptoms. All those carrying the DF508/DF508 genotype had pancreatic insufficiency. Pancreatic sufficiency was high and observed in about 1/3 of the remaining patients. There was an association between sweat test values (classified as <40 mEq/l, 40-60 mEq/l and >60 mEq/l) and genotypes in 48 individuals from our sample. A high proportion of individuals carrying genotypes with 1 or 2 DF508 mutations (DF508/DF508 6/6, DF508/other 13/16 and DF508/unknown 7/9) had Cl- values higher than 60 mEq/l. In contrast, few individuals carrying non-DF508 genotypes such as other/other (3/6) and other/unknown (3/10) mutations, had high sweat chloride levels (>60 mEq/l) (c2 = 12.0; 1 d.f.; P<0.001). DISCUSSION CF is described as a genetic disease characterized by three major features; chronic obstructive pulmonary disease, pancreatic insufficiency and abnormally high sweat electrolyte levels. The number of patients evaluated in this study is not sufficient to obtain a statistically significant estimate of the clinical aspects of the disease. However, our study population appears to be different from other CF populations. The classical triad of CF is not always present at the time of diagnosis. At that time, (14/52) of our patients did not need pancreatic enzyme supplements and were classified as pancreatic sufficient. This value is significantly higher than that obtained for other populations such as the U.S. (U.S. Cystic Fibrosis Foundation, 1997), where the reported proportion of registered patients who did not require enzymatic supplements was 17%. We detected a significant number of patients (12/48) with borderline sweat chloride (SWCl-) test values. On the other hand, the SWCl- distribution seems to be influenced by the types of mutations and genotypes (all the DF508/DF508 patients had high SWCl- values, while those with other genotypes had lower SWCl- values). As was observed in other populations, almost all the patients (49/52) had mild to severe pulmonary disease. These results suggest that our patients could represent a specific clinical population group, as proposed by the Cystic Fibrosis Foundation Consensus Panel (Rosenstein and Cutting, 1998). More studies are needed to confirm borderline sweat electrolyte levels and pancreatic insufficiency as a clinical entity. Uruguay has one of the highest health care standards among Latin American countries, 92.5% of the population having access to health care (PNUD, 1997). The country also has a low neonatal mortality (16.5/1000 live births, 1997) and a relatively low (1.7/1000) rate of deaths due to respiratory diseases. These data and the method of selection employed in this study suggest that bias due to early death without diagnosis would be unlikely in our sample. Patients with a typical clinical CF presentation and an incomplete genotype, but with several polymorphisms or variants other than IVS8-5T deserve further consideration. More research is necessary to elucidate the role played by variants in the etiology of this disease. It is possible that some mutations were undetected by the screening or mutation detection methods employed. This could be true for mutations located in introns or spanning multiple exons. The CF mutations and their relative frequencies observed in Uruguay are different from those described for other Latin countries. As in other predominantly Caucasian populations DF508 is the most frequent mutation, but its prevalence is relatively lower than in other populations, as it accounts for only 40% of the CF chromosomes. Various frequencies have been reported for the DF508 mutation in Latin American countries. It was 47-48.4% among Brazilians (Raskin et al., 1993; Bernardino et al., 2000) but significant interstate differences (27-53%) were observed (Raskin et al., 1993). Only 29.2% of the CF chromosomes in the Chilean population carry DF508 (Ríos et al., 1994), and the frequency of this mutation in the Mexican population is about 39% (Orozco et al., 1993). Finally, in Argentina, whose population is ethnically similar to Uruguay, 57% of the analyzed CF chromosomes carry the DF508 deletion (Chertkoff et al. 1997). These differences could be partially explained by variations in diagnostic criteria. For example, two positive sweat tests were required for the molecular analysis in the Argentine study, whereas borderline sweat chloride results were sufficient for analysis in Uruguay. We have also observed differences in the distribution and frequencies of non-DF508 mutations between Uruguayans and patients from other Latin American countries, in particular compared to the Argentinean population. Among Argentine CF patients, seven mutations (DF508, G542X, W1282X, N1303K, 17171G®A, R553X, R1162X) constituted 67.5% of the observed alleles (Chertkoff et al., 1997), while in our population 15 mutations corresponded to a similar cumulative percentage (71%). There is an agreement between the most common Uruguayan CFTR mutations (DF508, G542X, R1162X, N1303K, R334W, W1282X and R553X) and those reported in the geographical regions from where most Uruguayans’ ancestors originated, namely, Spain, the Canary Islands, Italy and the Basque regions. The Uruguayan population consists predominately of Caucasians, who immigrated to our country in several waves from different regions of Europe. Similar events occurred in the U.S. The overall CF incidence among Caucasians in the U.S. is 1/3300, with a 68% frequency of DF508 among CF chromosomes. However, the CF incidence in the U.S. ethnic Hispanic group is only 1/8-9000 and the prevalence of DF508 mutation in this group is only 46% (Cutting, 1997). These U.S. Hispanic population estimates are very similar to those obtained in Uruguay, with a CF incidence of 1/9600 and a frequency of 40% for the DF508 mutation. This similarity can be partially explained by a similar ethnic composition of Hispanics from the U.S. and the Uruguayan population (Caucasians, Amerindians and Africans) although the actual proportions are different. We have no information about the other mutations observed among Hispanics, which are only 4.9% of the total U.S. CF patients. Comparison of our mutation screening data with those from the U.S. Caucasian population, or with countries from where Uruguayan populations originated (Spain, France and Italy) indicates that the Uruguayan mutation spectrum is different. In U.S. CF patients, only three mutations (DF508, G542X, G551D) have gene frequencies higher than 2%, and altogether represent 72.5% of the CF chromosomes with a DF508 frequency of 68% (Cystic Fibrosis Foundation, 1997). The Argentine study shows similar proportions, with three mutations with a gene frequency exceeding 2% and covering 64% of the total number of chromosomes (DF508 = 57.0%, G542X = 3.94%, W1282X = 3.07%; Chertkoff et al., 1997). In Uruguay, however, seven mutations have a frequency of at least 2% and cover 59% of the tested CF chromosomes (Table 1). ACKNOWLEDGMENTS We thank Dr. Xavier Estivill for scientific support and kind help, allowing G. Luzardo to do part of this research in his laboratory. Research partially supported by the International Atomic Energy Agency (IAEA), Canadian Network of Centers of Excellence and Comisión Sectorial de Investigaciones Científicas (CSIC), Uruguay. REFERENCES Aznarez, I., Onay, T., Tzounzouris, J., Markiewicz, D., Mak, V., Tsui, L.C. and Zielenski, J. (1998). An efficient protocol for CFTR mutation detection based on multiplex heteroduplex analysis (mHET). Pediatr. Pulmonol. (Suppl.) 17: 332 (Abstract). Bernardino, A.L., Ferri, A., Passos-Bueno, M.R., Kim, C.E., Nakaie, C.M., Gomes, C., Damaceno, N. and Zatz, M. (2000). Molecular analysis on Brazilian cystic fibrosis patients reveals five novel mutations. Genet. Test 4: 69-74. Chertkoff, L., Visich, A., Bienvenu, T., Grenoville, M., Segal, E., Carniglia, L., Kaplan, J.C. and Barreiro, C. (1997). Spectrum of CFTR mutations in Argentine cystic fibrosis patients. Clin. Genet. 51: 43-47. Chillón, M., Casals, T., Mercier, B., Bassas, L., Lissens, W., Silver, S., Romey, M.C., Ruíz-Romero, J., Verlingue, C., Claustres, M., Nunes, V., Ferec, C. and Estivill, X. (1995). Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. New Eng. J. Med. 332: 1475-1480. Cutting, G.R. (1997). Genetic epidemiology and genotype/phenotype correlation’s. In: Program and Abstracts. NIH Consensus Development Conference on Genetic Testing for Cystic Fibrosis. April 14-16. Cystic Fibrosis Foundation (1997). Patient Registry 1996. Annual Data Report, Bethesda, MD, USA. Dörk, T., Macek Jr., M., Mekus, F., Tümmler, B., Tzountzouris, J., Casals, T., Krebsová, A., Koudová, M., Sakmaryová, I., Macek Sr., M., Vávrová, V., Zemková, D., Ginter, E., Petrova, N.V., Ivaschenko, T., Baranov, V., Witt, M., Pogorzelski, A., Bal, J., Zékanowsky, C., Wagner, W., Stuhrmann, M., Bauer, I., Seydewitz, H.H., Neumann, T., Jakubiczka, S., Kraus, C., Thamm, B., Nechiporenko, M., Livshits, L., Mosse, N., Tsukerman, G., Kadási, L., Ravnik-Glavac, M., Glavac, D., Komel, R., Vouk, K., Kucinskas, V., Krumina, A., Teder, M., Kocheva, S., Efremov, G.D., Onay, T., Kirdar, B., Malone, G., Schwarz, M., Zhou, Z., Friedman, K.J., Carles, S., Claustres, M., Bozon, D., Verlingue, C., Férec, C., Tzetis, M. ,Kanavakis, E., Cuppens, H., Bombieri, C., Pignatti, P.F., Sangiuolo, F., Jordanova, A., Kusic, J., Radojkovic, D., Sertic, J., Richter, D., Stavljenic Rukavina, A., Bjorck, E., Strandvik, B., Cardoso, H., Montgomery, M., Nakielna, B., Hughes, D., Estivill, X., Aznarez, I., Tullis, E., Tsui, L.-C. and Zielenski, J. (2000). Characterization of a novel 21-kilobase deletion, CFTRdele2,3 (21 kb), in the CFTR gene: a cystic fibrosis mutation of Slavic origin common in Central and East Europe. Hum. Genet. 106: 259-268. Fanen, P., Ghanem, N., Vidaud, M., Besmond, C., Martin, J., Costes, B., Plassa, F. and Goossens, M. (1992). Molecular characterization of cystic fibrosis: 16 novel mutations identified by analysis of whole cystic fibrosis conductance transmembrane regulator (CFTR) coding regions and splice site junctions. Genomics 13: 770-776. Gasparini, P., Nunes, V., Savoia, A., Dognini, M., Morral, N., Gaona, A., Bonizzato, A., Chillon, M., Sangiuolo, F., Novelli, G., Dallapiccola, B., Pignatti, P.F. and Estivill, X. (1991). The search for south European cystic fibrosis mutations: identification of two new mutations, four variants, and intronic sequences. Genomics 10: 193-200. Gibson, L.E. and Cooke, R.E. (1959). A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine iontophoresis. Pediatrics 23: 545-549. González, R. and Rodriguez, S. (1992). Guaraníes y Paisanos. Colección Nuestras Raíces. Ed. Nuestra Tierra, Montevideo, Uruguay. INE (Instituto Nacional de Estadística) (1996). VII Censo General de Población, Uruguay. Orita, M., Iwahara, H., Kanazawa, H., Hayashai, K. and Sekiya, T. (1989). Detection of polymorphisms on human DNA by gel electrophoresis as single strand conformation polymorphisms. Proc. Natl. Acad. Sci. USA 86: 2766-2770. Orozco, L., Salsedo, M., Lezana, J.L., Chavez, M., Valdez, H., Moreno, M. and Carnevale, A. (1993). Frequency of DF508 in a Mexican sample of cystic fibrosis patients. J. Med. Genet. 30: 501-502.Pi, R. and Vidart, D. (1969). El Legado de los Inmigrantes I y II. Ed. Nuestra Tierra, Montevideo, Uruguay. PNUD (1997). Human Development: Report 1997. Oxford University Press, New York, NY, USA, pp. 350. Raskin, S., Phillips III, J.A., Krishnamani, M.R.S., Vnencak-Jones, C., Parker, R.A., Rozov, T., Cardieri, J.M., Marostica, P., Abreu, F., Giugliani, R., Reis, F., Rosario, N.A., Ludwig, N. and Pilotto, R.F. (1993). DNA analysis of cystic fibrosis in Brazil by direct PCR amplification from guthrie cards. Am. J. Med. Genet. 46: 665-669. Ríos, J., Orellana, O., Aspillaga, M., Avedano, I., Largo, I. and Riveros, N. (1994). CFTR mutations in Chilean cystic fibrosis patients. Hum. Genet. 94: 291-294. Rosenstein, B.J. and Cutting, G.R. (1998). The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J. Pediatr. 132: 589-595. Sanger, F. and Coulson, A.R. (1975). A rapid method for determining sequences in DNA by primer synthesis with DNA polymerase. J. Mol. Biol. 94: 441-448. Sans, M. and Barreto, I. (1998). El problema de la integración de los negros a la sociedad general. In: Sociedad y Cultura en el Montevideo Colonial (Behares, L.E. and Cures, O., eds.). Recopilación de ponencias del seminario "Sociedad y Cultura en el Montevideo del Siglo XVIII". Cabildo de Montevideo, Uruguay. Sans, M., Salzano, F.M. and Chakraborty, R. (1997). Historical genetics in Uruguay: estimates of biological origins and their problems. Hum. Biol. 69: 161-170. The Cystic Fibrosis Genetic Analysis Consortium (1994). Population variations of common cystic fibrosis mutations. Hum. Mutat. 4: 167-177. Tsui, L.-C. (1992). The spectrum of cystic fibrosis mutations. Trends Genet. 8: 392-398. Valenzuela, C. (1988). Genética de la fibrosis quística. Rev. Chil. Pediatr. 31: 218-219. Welsh, M., Tsui, L.-C., Boat, T.F. and Beaudet, A.L. (1995). Cystic fibrosis, In: The Metabolic and Molecular Basis of Inherited Disease (Scriver, C.R., Beaudet, A.L., Sly, W.S. and Valle, D., eds.). 7th edn. McGraw Hill, New York, NY, USA, pp. 3799-3876. Zielenski, J. and Tsui, L.-C. (1995). Cystic fibrosis: genotypic and phenotypic variations. Annu. Rev. Genet. 29: 777-807. Zielenski, J., Bozon, D., Kerem, B., Markiewicz, D., Durie, P., Rommens, J. and Tsui, L.C. (1991). Identification of mutations in exon 1 through 8 of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene. Genomics 10: 229-235. |