![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
|
Rapid identification of single nucleotide polymorphisms by fluorescence-based capillary electrophoresis
ABSTRACT. We describe the application of two different fluorescence-based techniques (ddNTP primer extension and single-strand conformation polymorphism (SSCP)) to the detection of single nucleotide polymorphisms (SNPs) by capillary electrophoresis. The ddNTP primer extension technique is based on the extension, in the presence of fluorescence-labeled dideoxy nucleotides (ddNTP, terminators), of an unlabeled oligonucleotide primer that binds to the complementary template immediately adjacent to the mutant nucleotide position. Given that there are no unlabeled dNTPs, a single ddNTP is added to its 3’ end, resulting in a fluorescence-labeled primer extension product which is readily separated by capillary electrophoresis. On the other hand, the non-radioisotopic version of SSCP established in this study uses fluorescent dye to label the PCR products, which are also analyzed by capillary electrophoresis. These procedures were used to identify a well-defined SNP in exon 7 of the human p53 gene in DNA samples isolated from two human cell lines (CEM and THP-1 cells). The results revealed a heterozygous single-base transition (G to A) at nucleotide position 14071 in CEM cells, proving that both fluorescence-based ddNTP primer extension and SSCP are rapid, simple, robust, specific and with no ambiguity in interpretation for the detection of well-defined SNPs. Key words: Single nucleotide polymorphisms, Fluorescence-based techniques, THP-1 cells, CEM cells, p53 gene
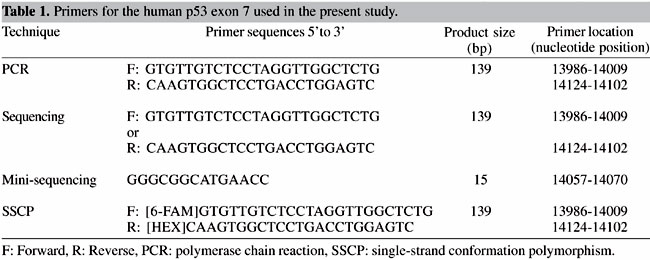
INTRODUCTION Single nucleotide polymorphisms (SNPs) are places along the chromosomes where the genetic code tends to vary from one person to another by just a single base (Nollou and Wagener, 1997). They are estimated to occur about once every 1000 bases along the 3-billion-base human genome (Nollou and Wagener, 1997). SNPs in genes or control regions may influence susceptibility to common diseases. Others probably have no function but could provide valuable markers for gene hunters; if they lie close to a susceptibility gene, they are likely to be inherited along with it. DNA sequencing is the method of choice to identify SNPs (Nollou and Wagener, 1997). However, DNA sequencing is in most cases a cost- and time-consuming task. To overcome this, we describe here the application of two different fluorescence-based techniques (dideoxy nucleotide, ddNTP primer extension and single-strand conformation polymorphism, SSCP) for the detection of SNPs by capillary electrophoresis. The ddNTP primer extension or minisequencing technique is based on the dideoxy single nucleotide extension of an unlabeled oligonucleotide primer. Briefly, an oligonucleotide primer is designed to bind to the complementary template immediately adjacent to the mutant nucleotide position (Figure 1A). When submitted to thermal cycling in the presence of DNA polymerase and fluorescence-labeled ddNTPs (terminators), the primer is extended one nucleotide. Given that there are no unlabeled dNTPs, a single ddNTP is added to its 3’ end resulting in a fluorescence-labeled primer extension product, which is readily separated by capillary electrophoresis and analyzed by an appropriate DNA fragment analysis software. Each individual ddNTP is assigned to a different fluorescent dye and therefore yields a different peak color of analyzed data. When a position demonstrates a heterozygous status, it is evidenced by the appearance of two peaks of different color. SSCP analysis is a widely accepted and useful application for detecting mutations or DNA polymorphisms based upon the ability of a single (or multiple) nucleotide change to alter the electrophoretic mobility of a single-stranded DNA molecule under non-denaturing conditions (Orita et al., 1989). The non-radioisotopic version of SSCP established in this study uses fluorescent dye to label the PCR products, which are then analyzed by capillary electrophoresis. In this case, a heterozygous position shows two peaks of different mobility. To illustrate the applicability of these techniques to the screening of DNA mutations or polymorphisms, we have selected a well-defined SNP in the human p53 gene. The p53 protein is a checkpoint control of the cell cycle and induces cell growth arrest and apoptosis (Cheng and Haas, 1990; Zhou et al, 1995). Furthermore, alteration of the p53 gene is strongly involved in the promotion of tumors. Detection of genetic alterations in p53 is therefore clinically relevant for diagnosis and treatment of various types of tumors. In this study, we analyzed genomic DNA isolated from the human lymphoblastic leukemia cell line CEM, which has been proven to harbor well-defined SNPs of the p53 gene. In these cells we specifically examined a G to A transition at position 14071 of codon 248 in exon 7 of the p53 gene (Cheng and Haas, 1990; Zhou et al, 1995). Another hematopoietic cell line, human acute monocytic leukemia THP-1 cells, served as control. MATERIAL AND METHODS CEM and THP-1 cells were maintained at 37°C with a 5% CO2 atmosphere and grown in RPMI supplemented with 10% fetal bovine serum (FBS) and 2 mM L-glutamine. Cells were lysed in 10 mM Tris, pH 8.0, 2 mM EDTA, 4 mM NaCl and 1% SDS containing proteinase K (2 mg/ml) and 2 mM EDTA and incubated overnight at 37ºC. DNA was extracted with chloroform:isoamyl alcohol (24:1, v/v), precipitated in ethanol and resolubilized in 10 mM Tris-HCl, 0.2 mM EDTA, pH 7.5 (TE buffer). Resulting high-molecular weight DNA was quantified by UV-spectrophotometry. PCR was performed by using primers spanning exon 7 of the human p53 cDNA sequence (accession number U49788) (see Table 1). Primers were constructed by solid phase phosphoramidite chemistry in a 394 DNA/RNA synthesizer (PE Applied Biosystems, Foster City, CA, USA). PCR was carried out in a total volume of 25 µl, which contained 100 ng of DNA sample, 1x PCR buffer, dNTP mixture (0.2 mM), oligonucleotide primers (20 µM each), MgCl2 (0.75 mM) and 0.5 U Taq DNA polymerase. The tubes were placed in a DNA thermocycler and amplified for 35 cycles using the following conditions: denaturation at 94°C for 30 s, annealing at 55°C for 30 s and extension at 72°C for 30 s, with a final extension step at 72°C for 7 min. PCR products of the expected size of 139 bp were analyzed by electrophoresis and visualized in a 2% agarose gel. Samples were subsequently run in a 0.5% agarose-2.5% Nusieve gel and the bands cut and extracted from the agarose with a Qiaex II gel extraction kit (Qiagen, Hilden, Germany) and resuspended in distilled water before measuring their absorbance at 260 nm.
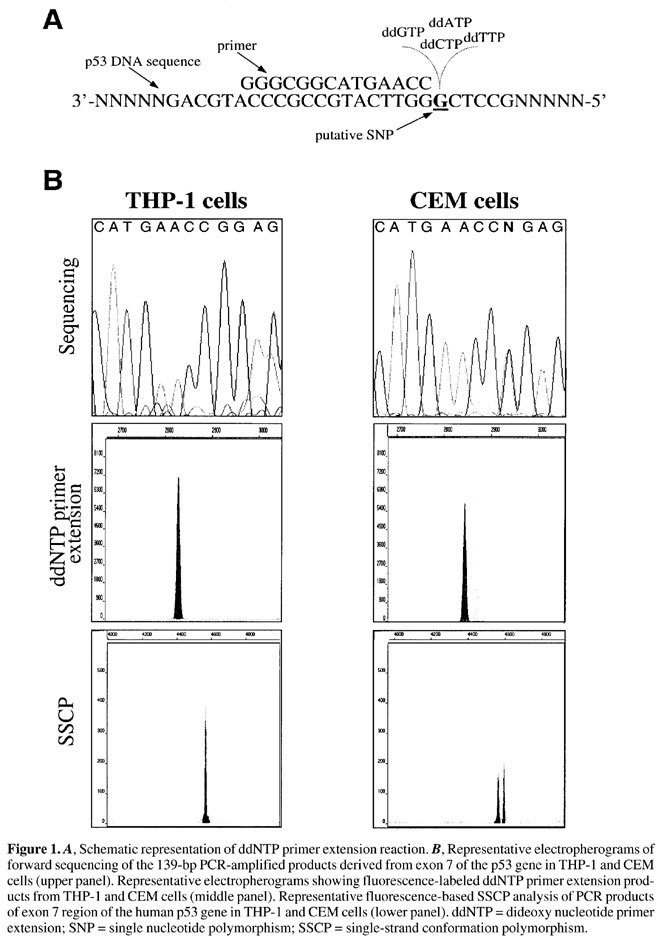
Sequencing of the PCR products was performed by automated laser-induced fluorescence DNA sequencing using the ABI Prism® Cycle dRhodamine terminator kit (PE Applied Biosystems). Briefly, reaction tubes (total volume 15 µl) containing 50-100 ng of DNA sample, 3.2 pmol of either the sense or the antisense oligonucleotides (see Table 1) and 7.1 µl of the sequencing mixture (Amplitaq® DNA Polymerase, FS with thermally stable pyrophosphatase), dNTPs, MgCl2 and ddNTPs cocktail (A-dye terminator labeled with dichloro[R6G], C-dye terminator labeled with dichloro[TAMRA], G-dye terminator labeled with dichloro[R110] and T-dye terminator labeled with dichloro[ROX]) in Tris-HCl buffer, pH 9.0, were placed in a DNA thermocycler and amplified at 96°C (30 s), 50°C (15 s) and 60°C (4 min) for 29 cycles. Sequencing reaction products were dried down and resuspended in 25 µl of template suppression reagent, denatured at 95°C for 2 min and then rapidly cooled on ice prior to loading into an automated ABI Prism® 310 Genetic Analyzer (PE Applied Biosystems). Sequencing reaction products were injected electrokinetically for 40 s at 2.0 kV and electrophoresed for 24 min at 15 kV, 9 µA at 50°C in a 47-cm length capillary using the performance optimum polymer 6 (POP6) with the laser set at a constant power of 9.9 mW. The resulting data were analyzed by a DNA Sequencing Analysis software (PE Applied Biosystems). Mini-sequencing was performed by automated laser-induced fluorescence DNA analysis using the ABI Prism® SNaPshotTM ddNTP primer Primer Extension kit (PE Applied Biosystems). ddNTP primer extension reactions were carried out in a total volume of 10 µl containing 0.15 pmol/µl of PCR-amplified DNA sample, 0.15 pmol/µl of oligonucleotide primer and 5 µl of SNaPshot ready reaction mixture containing Amplitaq® DNA Polymerase FS, A-dye terminator labeled with dichloro[R6G], C-dye terminator labeled with dichloro[TAMRA], G-dye terminator labeled with dichloro[R110] and T-dye terminator labeled with dichloro[ROX]. The oligonucleotide primer (5’-GGGCGGCATGAACC-3’) expanding from nucleotides 14057 to 14070 of a published human p53 cDNA sequence was designed to bind to the complementary template immediately adjacent to the expected SNP (nucleotide position 14071) (Table 1 and Figure 1A). The reaction tubes were placed in a DNA thermocycler and amplified for 25 cycles using the following conditions: denaturation at 96°C for 10 s, annealing at 58°C for 15 s and extension at 72°C for 30 s. The assay includes four positive and one negative control reactions. The four positive control reactions each contain 0.2 µg/µl of double-stranded DNA template (GEM -3Zf(+)) and a unique primer of different size and coding color (0.15 pmol/µl of 29-mer A-green; 0.75 pmol/µl of 34-mer T-red; 1.0 pmol/µl of 38-mer G-blue and 1.0 pmol/µl of 45-mer C-black). The negative control reaction includes a unique primer and no template DNA. The amplification conditions for control reactions were as follows: denaturation at 96°C for 10 s, annealing at 50°C for 5 s and extension at 60°C for 30 s for 25 cycles. ddNTP primer extension reaction products were analyzed with an automated ABI Prism® 310 Genetic Analyzer. Briefly, 1 µl of reaction product was mixed with 10 µl of deionized formamide and denatured at 95°C for 2 min and rapidly cooled on ice prior to loading into the ABI Prism® 310 Genetic Analyzer. Products were injected electrokinetically for 10 s at 15 kV and electrophoresed for 15 min at 15 kV, 9 µA, 60°C in a 47-cm length capillary using the performance optimum polymer 4 (POP4) with the laser set at a constant power of 9.9 mW. The resulting data were analyzed for peak color and fragment size using Genescan Analysis software (PE Applied Biosystems). Fluorescence-based SSCP analysis was performed on genomic DNA amplified with fluorescence-labeled PCR primers spanning exon 7 of the human p53 sequence (see Table 1). The specific exon 7 forward primer was labeled with 6-FAM (5-carboxyfluorescein) and the reverse primer with HEX (4,7,2’,4’,5’,7’-hexachloro-6-carboxyfluorescein). PCR was carried out as described above (see DNA amplification by PCR) and its efficiency was monitored by running an aliquot (3 µl) of the PCR product in a 2.5% agarose gel. Fluorescence-labeled PCR samples, without prior purification, were diluted 1:10 in deionized water. For capillary electrophoresis analysis, 1 µl of the diluted sample was combined with 10.5 µl deionized formamide, 0.5 µl 0.3 M NaOH and 0.5 µl of TAMRA-labeled GeneScan 350 size standard. The sample was denatured for 2 min at 96°C and subsequently cooled down to 4°C on ice prior to loading into the ABI Prism® 310 Genetic Analyzer. Separations were performed in either 3% or 5% GeneScan Polymer (GSP) in 10% glycerol (w/w) in 1x TBE. Samples were injected electrokinetically for 10 s at 15 kV and electrophoresed for 24 min at 13 kV, 10 µA, 44°C in a 47-cm length capillary with the laser set at a constant power of 9.0 mW. Raw data were analyzed using GeneScan 350 size standard. The sample was denatured for 2 min at 96°C and subsequently cooled down to 4°C on ice prior to loading into the ABI Prism® 310 Genetic Analyzer. Separations were performed in either 3% or 5% GeneScan Polymer (GSP) in 10% glycerol (w/w) in 1x TBE. Samples were injected electrokinetically for 10 s at 15 kV and electrophoresed for 24 min at 13 kV, 10 µA, 44°C in a 47-cm length capillary with the laser set at a constant power of 9.0 mW. Raw data were analyzed using GeneScan Analysis software. RESULTS AND DISCUSSION PCR amplification of exon 7 of the human gene p53 yielded a 139-bp DNA product. Direct DNA sequencing of this product from CEM cells showed a heterozygous single-base transition (G to A) at nucleotide position 14071 in exon 7 of the human p53 gene as demonstrated by an even incorporation of G and A at approximately half the height of the homozygous position (Figure 1B, upper panel). In contrast, THP-1 cells showed the wild-type DNA sequence at this nucleotide position. These results were confirmed by reverse sequencing of the PCR fragment which denoted a C to T transition in the DNA fragment amplified from CEM cells (data not shown). Mini-sequencing of the control DNA template (GEM -3Zf(+)) using each of the different control primers gave green-, red-, blue- and black-labeled DNA fragments corresponding to the extension products of each of the control primers incorporating a ddATP, ddTTP, ddGTP or ddCTP terminator, respectively (data not shown). The size of each of these fragments was 30, 25, 39 and 46 nucleotides and their run times were 10.56 ± 0.19, 10.72 ± 0.07, 10.77 ± 0.14 and 11.0 ± 0.11 min, respectively. Data from the ddNTP primer extension assay agreed with that obtained through prior sequencing analysis of the p53 DNA fragment. Thus, when running the mini-sequencing reaction products from CEM cells on the ABI Prism® 310 Genetic Analyzer, a blue and green doublet peak, corresponding to the heterozygous SNP in the 14071 nucleotide position of the human p53 gene, was observed (Figure 1B, middle panel). The size of both extension products was 15 nucleotides and the run times for the blue and green peaks were 10.13 ± 0.16 and 10.23 ± 0.17 min, respectively. Mini-sequencing of the DNA fragment from THP-1 cells revealed the presence of a single blue peak corresponding to the primer extension product (Figure 1B, middle panel). The size of the product was 15 nucleotides and the run time was 10.3 ± 0.27 min. Mini-sequencing of the DNA fragment from calibration curves indicated that the amount of mini-sequencing products to be injected can be as low as 1 µl (data not shown). Turnaround time in the present system is about 22 min per sample, including capillary filling, sample loading and separation. Thus, the maximum number of samples analyzable by the 310 Genetic Analyzer machine is 66 per day. This figure could actually be increased by multiplexing the mini-sequencing reaction by combining primers of different size with distinct dyes. PCR amplification of exon 7 of the human p53 gene with the fluorescence-labeled primers described in Table 1, yielded a 149-bp DNA product. Capillary electrophoresis SSCP analysis of the fluorescence-labeled DNA fragment from CEM cells revealed the presence of a blue doublet peak, corresponding to the heterozygous SNP (Figure 1B, lower panel). In contrast, SSCP analysis of the DNA fragment from THP-1 cells showed a single blue peak, the height of which was twice that obtained from CEM cells (Figure 1B, lower panel). Although qualitatively similar results were obtained from runs performed in 3% GSP and in 5% GSP, the highest concentration provided better peak resolution. The main advantages of using the 310 Genetic analyzer for SSCP analysis are i) the possibility to automate electrophoresis of the same sample at different temperatures, which maximizes the chances of detecting DNA alterations, ii) the elimination of radioactivity, iii) the possibility to perform rapid separations (fragments that are 300 bp or less in length can be separated in under 30 min), and thus iv) the ability to analyze a large number of samples. In summary, fluorescence-based ddNTP primer extension and SSCP are simple and versatile techniques for the detection of well-defined SNPs, such as those located in exon 7 of the human p53 gene. Both techniques have proven to be robust, specific and with no ambiguity
in interpretation, and it is likely that their implementation in laboratory practice will actually expand the potential applicability of fluorescence-based capillary electrophoresis technology to the analysis of DNA mutations or polymorphisms. ACKNOWLEDGMENTS Research supported in part by the Spanish Ministerio de Ciencia y Tecnología (SAF 00/0043). E. Titos is the recipient of a fellowship from Spain’s Ministerio de Sanidad y Consumo (BEFI 98/9314). REFERENCES Cheng, J. and Haas, M. (1990). Frequent mutations in the p53 tumor suppressor gene in human leukemia T-cell lines. Mol. Cell. Biol. 10: 5502-5509. Nollou, P. and Wagener, C. (1997). Methods for detection of point mutations: performance and quality assessment. Clin. Chem. 43: 1114-1128. Orita, M., Suzuki, Y., Sekiya, T. and Hayashi, K. (1989). Rapid and sensitive detection of point mutations and DNA polymorphisms using the polymerase chain reaction. Genomics 5: 874-879. Zhou, M., Yeager, A.M., Smith, S.D. and Findley, H.W. (1995). Overexpression of the MDM2 gene by childhood acute lymphoblastic leukemia cells expressing the wild-type p53 gene. Blood 85: 1608-1614. |