![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
ABSTRACT. Turner syndrome is caused by haploinsufficiency of the short arm of X-chromosome, and is usually diagnosed by karyotyping. This procedure is time-consuming, expensive and unfeasible for population screening. We propose molecular detection of 45XO Turner patients based on the ability of HpaII, a methylation sensitive endonuclease, to induce the cleavage of non-methylated DNA in the active X-allele. Genomic DNA was obtained from 22 patients with Turner syndrome confirmed by karyotype (45XO, N = 18; 45XO/46XX, N = 4). After digestion, DNA was amplified with primers directed to exon 1 of the androgen receptor (AR) gene and to the GAPDH control gene. Normal control females or mosaic patients, with a second methylated X-chromosome, escaped from HpaII digestion and produced a band corresponding to AR gene amplification. 45XO patients have just one active non-methylated X-chromosome, completely digested by HpaII, thus preventing the amplification of the AR gene. Three of the 45XO cases gave amplified bands, suggesting low-frequency mosaicisms that are not detected by karyotyping. Compared to classical karyotype studies for the detection of 45XO Turner patients, this new molecular method is simpler, faster and less expensive. Key words: Molecular diagnosis, Turner syndrome, X-Chromosome methylation
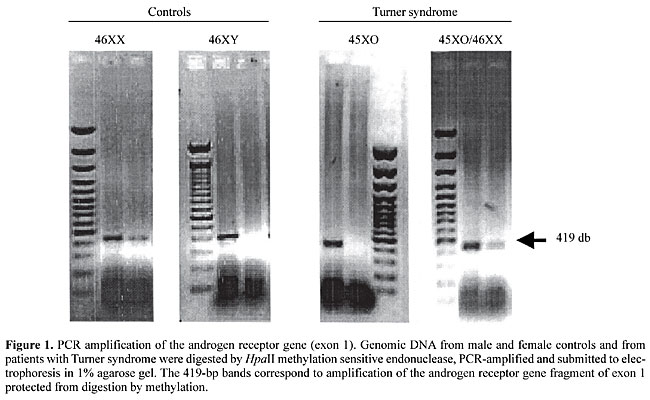
INTRODUCTION Turner syndrome (TS) is a chromosomal abnormality affecting one in 2000 live-birth girls (Hall and Gilchrist, 1990). This syndrome is characterized by clinical signs, including short stature, premature ovary failure and several somatic manifestations, which can be divided into three categories: i) skeletal anomalies, such as short metacarpals, Madelung’s deformity, cubitus valgo, short neck and cleft palate; ii) soft-tissue abnormalities related to lymphatic obstruction and consequent lymphedema, ungueal dysplasia, webbed neck, low implantation of the hair line; iii) visceral anomalies, including disorders of renal rotation, pielo-calicial duplicity and horse-shoe kidney; cardiac congenital defects, such as bicuspid aortic valve and aortic coarctation (Ogata et al., 2001; Sybert, 1998). TS has a wide spectrum of phenotypic manifestations, with short stature being the most common and sometimes the only clinical sign. Stature is partially related to the severity of X-chromosome structural alteration. X-chromosome monosomy (45XO karyotype) accounts for approximately 55% of TS patients, representing only 1% of all TS fetuses capable of surviving throughout gestation to term. Additionally, X-chromosome monosomy is the cause of 10% of all spontaneous abortions. Mosaicism, represented by 45XO/46XX or 45XO/47XXX cell lineages, has been detected in 30% of TS cases, and is usually caused by paternal meiotic non-disjunction. Structural abnormalities of the X-chromosome are observed in 15% of TS patients, characterized by only one functioning short arm. The most common structural aberrations are the long arm isochromosome (46XiXq), the short arm deletion (46XdelX) and the X-ring chromosome (46XrX) (Palmer and Reichmann, 1976). The severity of clinical manifestation is in part related to the type of chromosomal abnormalities, the time at which chromosome disjunction failed and the proportion of compromised cells in each tissue (Saenger et al., 2001). Genes related to TS are located in the short arm of the X-chromosome, and are able to escape methylation, determining the expression of both alleles (Kosho et al., 1999). X-chromosome methylation is the most important mechanism to silent the second allele, ensuring adequate gene dosage (Jang and Mao, 2000; Oakeley, 1999). Therefore, 45XO cells develop abnormal parity of sexual chromosomes during meiosis and functioning haploinsufficiency of genes located in the short arm. Several genes related to the clinical signs observed in TS patients have been mapped to this region, including short stature-related genes (Xp11.2-p22.1), ovary function regulatory genes (Xq13-q16) and genes related to lymphatic anomalies (Xp and Yp pseudoautosomal region) (Zinn and Ross, 2001). Karyotyping has been the method of choice to diagnose TS. It includes cell culture, which is one of the disadvantages of this technique, increasing the final cost, delaying the results and frequently determining a need for further blood samples. These intrinsic methodological limitations often prevent the use of karyotyping as a routine test to investigate patients with short stature or abortions suspected to be caused by TS. The high frequency of TS girls with short stature, the large spectrum of clinical manifestations, and the intrinsic limitations of karyotyping, compelled us to evaluate a simpler and faster molecular analysis to diagnose 45XO karyotype TS patients. PATIENTS AND METHODS Twenty-two TS cases confirmed by karyotyping were studied, including 45XO (N = 18) and 45XO/46XX (N = 4). We did not include 46XiXq or 46XrX TS patients. Genomic DNA was obtained from peripheral blood samples. After extraction, 1 mg of DNA was digested with the enzyme, HpaII (Boehringer Mannhein Co., Indianapolis, IN, USA) for 4 h at 37°C in order to induce DNA fragmentation in non-methylated regions. After digestion, the HpaII endonuclease was inactivated at 75°C for 15 min. Digested DNA was submitted to a PCR reaction, employing a pair of primers specific for the amplification of part of exon 1 of the androgen receptor (AR) gene (accession number GDB: 120556; OMIM entry: *313700) located in the long arm of the X-chromosome (region Xq11-q12). The forward and reverse primers were 5’-CTG AGC AAG AGA AGG GGA GGC GGG GTA AGG GAA GTA GGT GGA-3’ and 5’-CGA CTG CGG CTG TGA AGG TTG CTG TTC CTC ATC CAG-3’, respectively. All the experiments were performed in duplicate. Control experiments for the digestion step were developed using DNA and buffer without HpaII endonuclease. DNA quality after digestion was controlled by amplification of the housekeeping GAPDH-pseudogene, using forward primer 5’-TCT CCA GAA CAT CAT CCC TGC CAC-3’ and reverse primer 5’-TGG GCC ATG AGG TCC ACC ACC CTG-3’. The PCR reaction was made with digested DNA, 200 pmol of each deoxynucleotide, DMSO at a final concentration of 5%, 0.5 U of TaqDNA polymerase, 10X buffer and ddH2O to a final volume of 30 ml in a thermalcycler (GeneAmp PCR System 9700, Applied Biosystems, Forter City, CA, USA). After an initial denaturation at 95°C for 5 min, the samples underwent 35 cycles of 95°C for 1 min, 58°C for 3 min and 72°C for 3 min, followed by a prolonged extension at 72°C for 10 min. A buffer (3 ml of 10X Blue Juice Gel Loading Buffer, Life Technologies, Rockville, MD, USA) was added to the PCR product and submitted to electrophoresis in a 1% agarose gel, containing ethidium bromide. After electrophoresis, the bands were identified under UV light and the image captured by a digital camera. INTERPRETATION OF THE RESULTS The HpaII endonuclease is able to recognize and digest the CCGG sequences in which the second cytosine is not methylated. These non-methylated sequences are found in the active allele of 46XX normal individuals, or in those TS-45XO patients harboring only one functioning allele. Therefore, 45XO patients have one non-methylated active X-chromosome that can be fragmented by the HpaII enzyme. This specific fragmentation also occurs in the AR gene at the level of exon 1, a region containing two CCGG sequences. DNA digestion can prevent the amplification of this region during the PCR reaction. Conversely, the segment of the GAPDH-pseudogene, selected as a control, does not have the CCGG sequence and therefore is not digested by the HpaII enzyme.
Patients with 46XX or 45XO/46XX have a second methylated X-chromosome, protected from HpaII digestion, allowing the identification of one 419-bp band, corresponding to the amplification of a selected segment of exon 1 of the AR gene. RESULTS Amplification of the band corresponding to the GAPDH-pseudogene was observed in all cases, confirming DNA quality after digestion. Molecular analysis of 45XO karyotype TS patients (N = 18) gave two different patterns of amplification. In agreement with the karyotype, in 15/18 cases the band corresponding to the AR gene was not detected, indicating that only one active non-methylated X-chromosome was present. Unexpectedly, 3/18 TS patients with the 45XO karyotype repeatedly showed a band indicative of AR gene amplification, suggesting the presence of a 46XX cell lineage in a low frequency mosaicism not detected among the usual number of cells counted in a conventional karyotype. As expected, molecular analysis of TS patients 45XO/46XX was able to recognize the band resulting from AR gene amplification, confirming the presence of a second methylated X-chromosome in part of the cells. DISCUSSION Turner syndrome is one of the most frequent chromosomal abnormalities, associated with a variable spectrum of clinical manifestations. It requires strong clinical manifestations for adequate identification. Frequently, TS cases are unrecognized or have a delayed diagnosis during abnormal pubertal development or adult fertility impairment. Delayed recognition provokes suboptimal therapy, with permanent undesirable consequences. Upon clinical suspicion, karyotyping has been the method of choice to confirm the diagnose of TS. Intrinsic characteristics of karyotyping generates high costs and several practical limitations, preventing its use as a routine screening test for high-risk groups, such as girls with short stature. We describe the use of the methylation-sensitive enzyme HpaII, an endonuclease capable of recognition and digestion of non-methylated CCGG sequences, allowing the identification of individuals with only one X-chromosome. When compared to classical karyotyping, this is a simpler and faster method for molecular detection of 45XO TS patients. The low cost and the simplified characteristics of this new molecular method opens the possibility of screening patients at high risk for TS, such as short girls. In agreement with a previous report (Yorifuji et al., 1997), high sensitivity in the recognition of low-frequency XX cell lineage in patients with mosaicism was also observed. However, this method has the disadvantage of its limitation to detect other chromosome abnormalities that sometimes cause TS. The concomitant use of primers directed towards other methylated genes located in the short arm of X-chromosome can potentially overcome these limitations. We recommend that these tests be performed in duplicate and that they include negative controls, in order to detect failures related to insufficient digestion or inadequate amplification. The PCR amplification of a housekeeping gene, such as the GAPDH gene, is essential to ensure the quality of the DNA after digestion.We conclude that molecular detection of 45XO patients with Turner syndrome can be easily and efficiently employed, representing an important method for molecular screening of these patients in high-risk groups. REFERENCES Hall, J.G. and Gilchrist, D.M. (1990). Turner syndrome and its variants. Pediatr. Clin. North Am. 37: 1421-1436. Jang, S.L. and Mao, L. (2000). Methylation patterns in human androgen receptor gene and clonality analysis. Cancer Res. 60: 864-866. Kosho, T., Muroya, K., Nagai, T., Fujimoto, M., Yokoya, S., Sakamoto, H., Hirano, T., Terasaki, H., Ohashi, H., Nishimura, G., Sato, S., Matsuo, N. and Ogota, T. (1999). Skeletal features and growth patterns in 14 patients with haploinsufficiency of SHOX: implications for the development of Turner syndrome. J. Clin. Endocrinol. Metab. 84: 4613-4621. Oakeley, E.J. (1999). DNA methylation analysis: a review of current methodologies. Pharmacol. Ther. 84: 389-400. Ogata, T., Muroya, K., Matsuo, N., Shinohara, O., Yorifuji, T., Nishi, Y., Hasegawa, R., Horikawa, R., and Tachibana, K. (2001). Turner syndrome and Xp deletions: clinical and molecular studies in 47 patients. J. Clin. Endocrinol. Metab. 86: 5498-5508. Palmer, C.G. and Reichmann, A. (1976). Chromosomal and clinical findings in 110 females with Turner syndrome. Hum. Genet. 35: 35-49. Saenger, P., Wikland, K.A., Conway, G.S., Davenport, M., Gravholt, C.H., Hintz, R., Hovatta, O., Hultcrantz, M., Landin-Wilhelmsen, K., Lin, A., Lippe, B., Pasquino, A.M., Ranke, M.B., Rosenfeld, R. and Silberbach, M. ( 2001). Recommendations for the diagnosis and management of Turner syndrome. (Fifth International Symposium on Turner Syndrome). J. Clin. Endocrinol. Metab. 86: 3061-3069. Sybert, V.P. (1998). Cardiovascular malformations and complications in Turner syndrome. Pediatrics 101: E11. Yorifuji, T., Muroi, J., Kawai, M., Sasaki, H., Momoi, T. and Furusho, K. (1997). PCR-based detection of mosaicism in Turner syndrome patients. Hum. Genet. 99: 62-65. Zinn, A.R. and Ross, J.L. (2001). Molecular analysis of genes on Xp controlling Turner syndrome and premature ovarian failure. Semin. Reprod. Med. 19: 141-146. |