![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
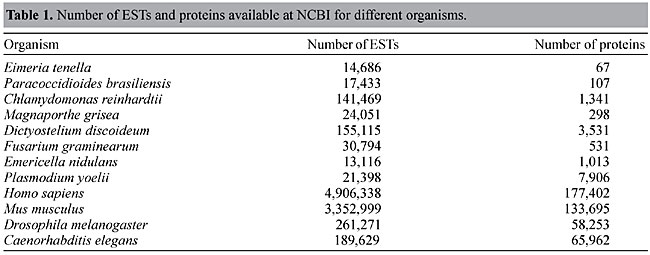
ABSTRACT. Microorganisms with large genomes are commonly the subjects of single-round partial sequencing of cDNA, generating expressed sequence tags (ESTs). Usually there is a great distance between gene discovery by EST projects and submission of amino acid sequences to public databases. We analyzed the relationship between available ESTs and protein sequences and used the sequences available in the secondary database, clusters of orthologous groups (COG), to investigate ESTs from eight microorganisms of medical and/or economic relevance, selecting for candidate ESTs that may be further pursued for protein characterization. The organisms chosen were Paracoccidioides brasiliensis, Dictyostelium discoideum, Fusarium graminearum, Plasmodium yoelii, Magnaporthe grisea, Emericella nidulans, Chlamydomonas reinhardtii and Eimeria tenella, which have more than 10,000 ESTs available in dbEST. A total of 77,114 protein sequences from COG were used, corresponding to 3,201 distinct genes. At least 212 of these were capable of identifying candidate ESTs for further studies (E. tenella). This number was extended to over 700 candidate ESTs (C. reinhardtii, F. graminearum). Remarkably, even the organism that presents the highest number of ESTs corresponding to known proteins, P. yoelii, showed a considerable number of candidate ESTs for protein characterization (477). For some organisms, such as P. brasiliensis, M. grisea and F. graminearum, bioinformatics has allowed for automatic annotation of up to about 20% of the ESTs that did not correspond to proteins already characterized in the organism. In conclusion, 4093 ESTs from these eight organisms that are homologous to COG genes were selected as candidates for protein characterization. Key words: Expressed sequence tag (EST), Protein characterization, Cluster of orthologous groups, Microorganism genomes INTRODUCTION During the last decade, countless genome projects have been developed and the study of microorganism genomes, especially pathogenic ones, has undergone significant advances. The availability of genome sequences and other large genetic datasets of many organisms will lead to a revolution in the understanding of the complex biochemical machinery of the cells and the treatment of genetic and infectious diseases. However, gene discovery has not been followed, in the same proportion, by protein deposits in databases, especially when the microorganism has a large genome and the characterization of its gene products is carried out by the characterization of the transcriptome and the proteome. Since the main target for drug development are proteins, the identification and characterization of them have attracted considerable attention from researchers outside the genome initiatives. Research on protein characterization has benefited profoundly from transcriptome and proteome projects, as well as from the development of bioinformatics tools and secondary biological databases (where sequences are organized in categories). Transcriptome projects provide important clues for understanding the structure and organization of biological systems, allowing in many cases the identification of genes that are over-expressed or silenced in certain tissues or organisms. In many transcriptome projects the production of expressed sequence tags (ESTs) has been the main resource for gene discovery. ESTs are short sequences (~300 nucleotides), generated from the end of cDNAs randomly selected from a given library (Adams et al., 1991). Since the cDNAs are generated from mRNAs produced by a cell at a given moment, ESTs represent the expression profile of a tissue or organism. Although the protocols to produce ESTs present some limitations such as a high error rate and a large number of chimera sequences (Boguski et al., 1993; Wolfsberg and Landsman, 1997), ESTs are very informative, generating a large amount of information in a short span of time. Thus, a large number of ESTs have been published in public databases, constituting an extensive resource for several purposes such as mapping, functional studies and the starting point for protein characterization. The EST collection at the National Center for Biotechnology Information (NCBI - USA) for example, contains ESTs from 470 different organisms (December, 2002), from Homo sapiens to protozoa (www.ncbi.nlm.nih.gov/dbEST/), adding up to more than 15,000,000 sequences. Homology searches using bioinformatics tools, such as BLAST (Altschul et al., 1997), are used in the identification of ESTs. However, there has not been a systematic approach that uses known proteins, classified in secondary databases, to investigate ESTs generated by high throughput projects. Such proteins can be grouped by virtue of their sequence similarity, functional association, or spatial-temporal distribution, and this constitutes important information to investigate their function. One group of such proteins that is available in public databases is the sequence set COG (clusters of orthologous groups - NCBI). This sequence set is a database with phylogenetic classification of proteins encoded in complete genomes (bacteria, yeast, and includes Drosophila melanogaster and Caenorhabditis elegans) (Tatusov et al., 2001). Given the great distance between the proteome and transcriptome discoveries, we propose to use the sequences available in COG to investigate ESTs from eight model organisms of medical and/or economic relevance, selecting for candidate ESTs that may be further pursued for protein characterization. The organisms we chose are Paracoccidioides brasiliensis, Dictyostelium discoideum, Fusarium graminearum, Plasmodium yoelii, Magnaporthe grisea, Emericella nidulans, Chlamydomonas reinhardtii, Eimeria tenella, which have more than 10,000 ESTs available in dbEST (NCBI EST database, December 2002). Paracoccidioides brasiliensis is a dimorphic pathogenic fungus that infects people living in South America, causing systemic mycosis (Cano et al., 1998). It is acquired by inhalation of spores from conidia present in the environment, whereas the pathogen’s morphology is a yeast form. Transformation from one form to the other occurs inside the host and is triggered by temperature (35-37°C) (Cano et al., 1998). Venancio et al. (2002) showed by differential display that some genes are differently expressed in each fungal phase. The study of gene expression and regulation of each phase of the fungus will be an aid to developing new therapies and treatment for patients with this type of infection. Dictyostelium discoideum is a soil-living amoeba, whose EST database at dbEST has already reached 155,032 submitted sequences. The cDNA, Genome and Proteome Project are in an integrated database (http://dictybase.org; Urushihara, 2002). These organisms have unique advantages for studies of fundamental cellular processes, such as cytokinesis, motility, phagocytosis, chemotaxis, signal transduction, etc. Since these cellular behaviors and biochemical mechanisms are either absent or are less accessible in other model organisms, D. discoideum was one of the model organisms chosen by the National Institutes of Health for functional analysis of sequenced genes (http://dictybase.org). Fusarium graminearum is the fungus that causes fusarium head blight in cereals. This fungus causes direct yield loss in cereals and also produces mycotoxins in the infected grain. The most common mycotoxin produced by F. graminearum is deoxynivalenol (DON). The fungus, F. graminearum, is now known to consist of eight separate genetic lineages, some of which are restricted to particular continents. These lineages may be distinguished by DNA sequence comparisons and have considerable variation in their ability to produce mycotoxins (O’Donnell et al., 2000). Plasmodium yoelii has been isolated from the blood of shiny thicket rats from the Central African Republic, from Brazzaville and from Western Nigeria. Three subspecies are recognized, P. yoelii yoelii, P. yoelii killicki and P. yoelii nigeriensis. This parasite is readily grown in laboratory mice and rats, where it shows a preference for immature red blood cells. Infections are asynchronous, with a periodicity of 22-25 h. Like P. berghei, the parasite may be transmitted in the laboratory by Anopheles stephensi mosquitoes (Carlton et al., 2002). Magnaporthe grisea is a fungus that infects rice (Grayer and Kokubun, 2001). Emericella nidulans is a filamentous fungus that produces penicillin (Brakhage, 1997). Chlamydomonas reinhardtii is a green unicellular alga that has been used as an experimental model organism for circadian rhythm research for more than 30 years (Werner, 2002), and it has emerged as a powerful model system for studying the biosynthesis of the photosynthetic apparatus (Rochaix, 2002). Eimeria tenella is one of the most important species of Eimeria spp., coccidial parasites that cause cecal coccidioises in chickens (Shirley, 2000). The analyses conducted in this work will enable one to infer the function of orthologous genes in the selected organisms using sequences from microorganisms present in COG, the genomes of which are complete and information about their known proteins is available and has been classified. This strategy could be used to investigate ESTs from any other organism. We show here that a considerable number of proteins have been sampled at the EST level. Thus, cDNA clones used to generate such ESTs are a resource for the characterization of proteins that constitute candidates to inclusion in the COG database. MATERIAL AND METHODS The number of ESTs and proteins for organisms bearing more than 10,000 ESTs available at NCBI was compared in order to study how well protein discovery follows EST discovery. Eight microorganisms were chosen based on their medical and/or economic relevance and on the small number of characterized proteins that they possess in comparison to ESTs. The sequences used in this study are available from public databases at NCBI - USA (http://ncbi.nlm.nih.gov - Jan/2003). ESTs from all organisms were downloaded from dbEST. Protein sequences from individual organisms were retrieved from Entrez Protein (Table 1) and 77,114 protein sequences were downloaded from the COG ftp site (ftp://ftp.ncbi.nih.gov/pub/COG).
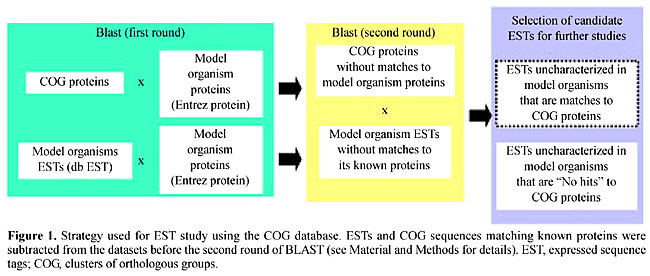
Homology searches were performed using BLAST (Altschul et al., 1997) with all default parameters. The SEG filter was used on the first and second rounds of BLAST, except for P. brasiliensis, E. nidulans and M. grisea on the second round. The EXPECT threshold was set to 10-10. Sequences from COG or ESTs that match protein sequences already identified in the microorganisms chosen for study were detected by BLAST (first round) and removed from the dataset for further analysis using Linux shell scripts. The remaining COG sequences were used in BLAST searches against the remaining ESTs (second round - Figure 1), using the parameters above. Results were parsed and inserted into a MySQL database. Data mining was performed and results grouped by organism.
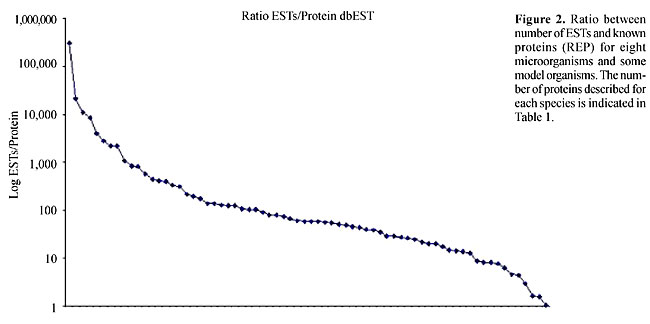
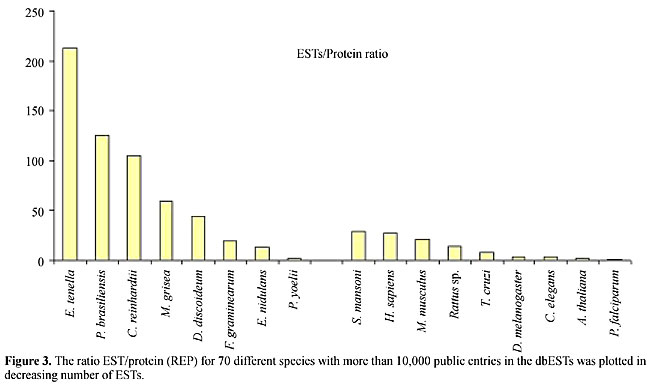
RESULTS Comparison of the number of ESTs and characterized proteins for each organism There is a great discrepancy between the number of ESTs that have been discovered and the number of proteins characterized for several organisms. Table 1 shows the number of ESTs and proteins available in public databases for the organisms used in this study as well as some other model organisms (for the number of ESTs for all organisms see http://www.ncbi.nlm.nih.gov/dbEST/index.html). Figure 2 shows the ratio of ESTs/proteins for several organisms. Figure 3 shows the results for organisms with more than 10,000 ESTs, adding up to 70 species. As can be seen, the ratio distribution is not homogenous and the discrepancy between the number of ESTs and proteins is particularly great for some groups, such as plants. For model organisms such as H. sapiens, Mus musculus, D. melanogaster and C. elegans, for example, the discrepancy is much smaller.
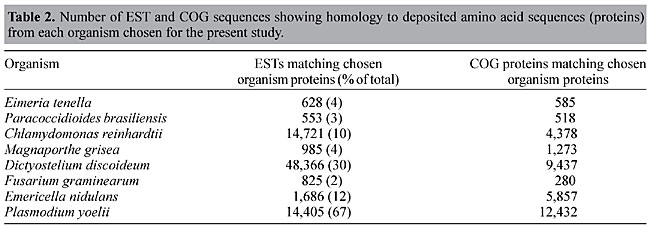
Homology searches Homology searches were performed in two rounds (see Material and Methods - Figure 1) to allow for screening of EST and COG sequences that already represent known proteins in each selected organism. First round BLAST searches were performed to screen for the COG or EST sequences that already have matches to known proteins of the chosen organism. As can be seen in Table 2, the percentage of ESTs that showed a match to known proteins varies from 2% (F. graminearum) to 67% (P. yoelii). These results probably reflect the different efforts in protein characterization conducted for each microorganism. Table 2 also shows the number of COG proteins that were removed for subsequent data processing.
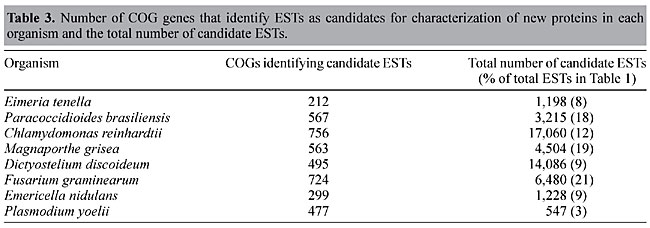
Second round The results of BLAST searches using COG proteins to select ESTs that correspond to unidentified proteins are presented in Table 3. A total of 77,114 protein sequences from COG were used, corresponding to 3,201 distinct genes. At least 212 (E. tenella) of these were capable of identifying candidate ESTs for further studies. This number extends to over 700 candidate ESTs (C. reinhardtii, F. graminearum). Remarkably, even the organism that presents the highest number of ESTs corresponding to known proteins, P. yoelii (67% - Table 2), shows a considerable number of candidate ESTs for further studies (477 - Table 3). A total of 4,093 ESTs were selected for all the organisms. Since an EST may not always contain the complete coding sequence, we decided to collect all possible candidate ESTs that show similarities to COG proteins (Table 3 - third column). Candidate ESTs may be used to characterize proteins of different Pathways or Cellular Function already cataloged in the COG database. This information is provided as complementary data in the server at Laboratório de Biodados, UFMG (http://biodados.icb.ufmg.br). A significant number (up to around 20% for three of the chosen organisms - Table 3) of the ESTs not related to proteins already described for the organism can be automatically annotated and classified into COG categories by the described procedure.
CONCLUSIONS The analysis of the proportion of ESTs versus proteins deposited in NCBI Taxonomy (or Entrez Protein) clearly demonstrates that there is a large deficit of characterized proteins compared to ESTs, particularly for some pathogenic microorganisms. For organisms such as H. sapiens or M. musculus this deficit is not so large, however. This is probably due to their relevance and historical or epidemiological scientific interest. Nevertheless, for some important microorganisms, such as P. brasiliensis, protein characterization is in behind since the technology of high throughput cDNA production has recently allowed for a large number of excellent quality reads in short time, and this has not yet been followed by extensive protein characterization. The relevance of such organisms, on the other hand, justifies the accurate and systematic analyses of EST databases in the quest for ESTs more relevant to study. We propose here a computational agent that will automatically search for ESTs that might represent a starting point for the characterization of proteins on demand for full-length cDNA sequencing. The relevance of the chosen secondary database, COG, is supported by the fact that included proteins are presented by three or more of the organisms with a complete genome; however, other secondary databases can be easily used by the agent that we propose. As shown in Table 3, even the most characterized set of ESTs, the one generated from P. yoelli, can be a subject for the proposed selection, generating candidate ESTs. For some organisms, such as P. brasiliensis, M. grisea and F. graminearum, the analysis performed here has allowed automatic annotation of up to 20% of the ESTs that do not correspond to proteins already characterized in each organism, thus providing a powerful auxiliary tool for annotation, allowing additionally the inclusion of such ESTs in functional gene categories. The advantage of inclusion of gene information at the protein level in addition to nucleotide information is seminal for the analysis of new genomes, since protein databases are often the subject of homology searches. However, protein to protein comparison is only reliable when protein sequences themselves are used, not ESTs. Protein characterization is therefore of great relevance for genomics. In this regard, the enrichment added by secondary protein databases, such as COG, has a remarkable impact, since these contain not only the raw sequences but also functional information. The computational agent proposed here constitutes an important tool for this purpose. Data mining is usually the final step in a gene discovery program. The concept behind this work could be understood as a reverse annotation, in the sense that the proteins of interest are chosen from secondary databases and automatic bioinformatics tool searches, with tBLASTn, for ESTs that fit as a target for annotation, as well as a candidate for protein characterization. Furthermore, the automation allowed by this process makes possible the processing of a large number of sequences from more than one organism in a short span of time. ACKNOWLEDGMENTS The Laboratório de Biodados of the Rede Genoma de Minas Gerais is supported by a grant from FAPEMIG and MCT/Brasil. This work includes concepts from A.C. Faria-Campos’ Ph.D thesis and results from a workshop held at the Departamento de Bioquímica e Imunologia, UFMG, attended by A.C. Faria-Campos, C. Anacleto, C.B. Carvalho (authors) and A.S. Barouni, C.D.S. Macêdo, F. Ribeiro, D.N. Santos and C.E. Calzavara-Silva, who contributed with suggestions. The authors are grateful to S.V.A. Campos, J.A. Tôrres (DCC - UFMG), F.C. Peixoto and O.S.F. Carvalho (LCC - CENAPAD/MG-CO) for their help with parsing and for providing additional workstations for data processing. The authors are also grateful to Dr. Darren Natale (PIR, Georgetown University Medical Center) for critically reviewing the manuscript. REFERENCES Adams, M.D., Kelley, J.M., Gocayne, J.D., Dubnick, M., Polymeropoulos, M.H., Xiao, H., Merril, C.R., Wu, A., Olde, B., Moreno, R.F. et al. (1991). Complementary DNA sequencing: expressed sequence tags and human genome project. Science 252: 1651-1656. Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W. and Lipman, D.J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. Boguski, M.S., Lowe, T.M. and Tolstoshev, C.M. (1993). dbEST- database for “expressed sequence tags”. Nat. Genet. 4: 332-333. Brakhage, A.A. (1997). Molecular regulation of penicillin biosynthesis in Aspergillus (Emericella) nidulans. FEMS Microbiol. Lett. 148: 1-10. Cano, M.I., Cisalpino, P.S., Galindo, I., Ramirez, J.L., Mortara, R.A. and da Silveira, J.F. (1998). Electrophoretic karyotypes and genome sizing of the pathogenic fungus Paracoccidioides brasiliensis. J. Clin. Microbiol. 36: 742-747. Carlton, J.M., Angiuoli, S.V., Suh, B.B., Kooij, T.W., Pertea, M., Silva, J.C., Ermolaeva, M.D., Allen, J.E., Selengut, J.D., Koo, H.L., Peterson, J.D., Pop, M., Kosack, D.S., Shumway, M.F., Bidwell, S.L., Shallom, S.J., van Aken, S.E., Riedmuller, S.B., Feldblyum, T.V., Cho, J.K., Quackenbush, J., Sedegah, M., Shoaibi, A., Cummings, L.M., Florens, L., Yates, J.R., Raine, J.D., Sinden, R.E., Harris, M.A., Cunningham, D.A., Preiser, P.R., Bergman, L.W., Vaidya, A.B., van Lin, L.H., Janse, C.J., Waters, A.P., Smith, H.O., White, O.R., Salzberg, S.L., Venter, J.C., Fraser, C.M., Hoffman, S.L., Gardner, M.J. and Carucci, D.J. (2002). Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature 419: 512-519. Grayer, R.J. and Kokubun, T. (2001). Plant-fungal interactions: the search for phytoalexins and other antifungal compounds from higher plants. Phytochemistry 56: 253-263. O’Donnell, K., Kistler, H.C., Tacke, B.K. and Casper, H.H. (2000). Gene genealogies reveal global phylogeographic structure and reproductive isolation among lineages of Fusarium graminearum, the fungus causing wheat scab. Proc. Natl. Acad. Sci. USA 97: 7905-7910. Rochaix, J.D. (2002). Chlamydomonas, a model system for studying the assembly and dynamics of photosynthetic complexes. FEBS Lett. 529: 34-38. Shirley, M.W. (2000). The genome of Eimeria spp., with special reference to Eimeria tenella - a coccidium from the chicken. Int. J. Parasitol. 30: 485-493. Tatusov, R.L., Natale, D.A., Garkavtsev, I.V., Tatusova, T.A., Shankavaram, U.T., Rao, B.S., Kiryutin, B., Galperin, M.Y., Fedorova, N.D. and Koonin, E.V. (2001). The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29: 22-28. Urushihara, H. (2002). Functional genomics of the social amoebae, Dictyostelium discoideum.Mol. Cells 13: 1-4. Venancio, E.J., Kyaw, C.M., Mello, C.V., Silva, S.P., Soares, C.M., Felipe, M.S. and Silva-Pereira, I. (2002). Identification of differentially expressed transcripts in the human pathogenic fungus Paracoccidioides brasiliensis by differential display. Med. Mycol. 40: 45-51. Werner, R. (2002). Chlamydomonas reinhardtii as a unicellular model for circadian rhythm analysis. Chronobiol. Int. 19: 325-343. Wolfsberg, T.G. and Landsman, D. (1997). A comparison of expressed sequence tags (ESTs) to human genomic sequences. Nucleic Acids Res. 25: 1626-1632. |
|