![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
ABSTRACT. We have identified a new mutant mouse that we have named new mouse neurological mutant 3 (NM3); it may be a useful model to understand the underlying molecular and genetic basis of Parkinson’s disease (PD). A mouse carrying the NM3 mutation arose spontaneously in an RIIIS/J breeding colony and was identified as having a movement disorder. Upon neurological examination of these mice, their movement was found to be slow and abnormal, with characteristic choreaform and bradykinetic-type movements, typical of PD. The importance of the gene mutation in NM3 in the molecular pathway involved in this pathology is underscored by the fact that these mice do not survive past weaning age if they are homozygous for the genetic mutation. We localized the gene mutation by positional cloning and genetic mapping to mouse chromosome 2 in an area that corresponds to human chromosome 2q24-31, which does not contain any known genes associated with PD. However, there was a significant decrease of 15-20% in the levels of dopamine, and its principal metabolite, 3,4-dihydroxyphenylacetic acid, in the midbrain of affected mice. Low concentrations of these substances are associated with PD in human patients, making these mutant mice candidates for studies of this disease. Key words: Parkinson’s disease, Mutant mouse, Dopamine INTRODUCTION Parkinson’s disease (PD) is one of the most common neurodegenerative disorders, primarily affecting voluntary, coordinated movement; it is characterized by akinesia, rigidity, and tremor at rest. The cause of death of dopaminergic neurons and subsequent dopamine depletion in the brain of patients with PD remains unknown, although it has been postulated that it is initiated by the interaction of genetic predisposition and environmental or endogenous nigral neurotoxins (Carlsson and Carlsson, 1990; Albin and Greenamyre, 1992; Fahn and Cohen, 1992; Beal et al., 1993; Tanner, 1994). Indeed, much of the recent research efforts have focused on the loss of dopaminergic neurons in the substantia nigra, although the true etiology of this disease is still unknown. Speculating that both environmental and genetic factors play an important role in the pathophysiology of PD, molecular genetic research is now poised to identify the defect(s) in this disorder, to elucidate inherited risk factors and ultimately identify the sequence of molecular events that leads to the prototypic pathology of PD. Genetic linkage and molecular cloning studies are of critical importance in clarifying the pathogenesis of disease processes. Finding and characterizing animal genetic models of disease now offers a hope towards our understanding of the complex etiology of human health issues like PD. In an effort to explore the environmental roots of PD, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) has been used as a potent neurotoxin to approximate, in animals, the pathology associated with human PD. Although not identical, MPTP neurotoxicity and PD are strikingly similar (Langston and Irwin, 1986). The symptomatology of each reflects a disruption of nigrostriatal pathways, with resting tremors, rigidity, and bradykinesia as features of both conditions. Mice treated with MPTP develop dopamine depletion, but the associated nigral cell death is not universally observed, with strain-related differences (Hallman et al., 1985; Willis and Donnan, 1987; Hamre et al., 1999). It has been suggested that the differences in neuronal cell loss between different strains of mice may, in fact, be related to differences in the functioning of the plasma membrane dopamine transporter (Bezard et al., 1999). Moreover, mutations in the a-synuclein gene are theorized to be associated with rare cases of autosomal-dominant PD, and neuronal cell death is not a feature of these mutant mice, with or without involvement of MPTP (Rathke-Hartlieb et al., 2001). Thus, the discovery of synthetic, as well as natural, molecules able to reproduce in the animals the biochemical and morphological alterations of neurodegenerative disorders has provided a major impetus towards an environmental hypothesis of neurodegeneration (Piccoli et al., 1992). While the environmental basis underlying PD has been vigorously studied, there have been few reports examining the predisposing genetic factors and/or molecular pathways that may contribute to PD. The field is now set to exploit new model systems in which to identify the gene(s) and molecular pathways underlying PD. A mouse carrying the neurological mutant 3 (NM3) mutation arose spontaneously in an RIIIS/J breeding colony and was identified as having a movement disorder. Upon neurological examination of these mice, their movement was found to be slow and abnormal, with characteristic choreaform and bradykinetic-type movements, typical of PD. We initiated a genetic study of these mice. MATERIAL AND METHODS Genetic mapping We initiated work on a high-resolution genetic and physical map to identify and characterize the genetic mutation in NM3. We utilized meiotic recombination to rapidly limit the genomic region in which the target gene must be located. Future completion of this step will provide positive identification of the NM3 gene and its mutation. To fine map, the segregation of the neurologic PD phenotype was examined in fully tested intercross progeny. Simple sequence repeat markers, which are often polymorphic among different inbred strains and highly polymorphic between wild-derived inbred (i.e., Cast/Ei) and other inbred strains, were utilized in the region of interest on chromosome 2 and were used to detect polymorphisms between RIIIS/J (the parent strain NM3 arose spontaneously from) and CAST/Ei (the strain to which NM3 has been intercrossed and backcrossed) DNA. Determination of dopamine and its metabolites in brain samples Brain samples were collected and frozen for tissue analysis of dopamine by HPLC according to the method of Champney et al. (1992). The brain regions were removed from the freezer, placed in an appropriate amount of ice-cold 0.16 M perchloric acid (PCA) containing dihydroxybenzylamine (DHBA, the amine internal standard, 200 ng/ml). For the midbrain, 100 ml PCA containing DHBA is appropriate for samples weighing between 10 and 20 mg. Other brain regions require approximately 100 ml PCA with DHBA per 10 mg wet weight. The tissue samples were disrupted by sonication and centrifuged at 13,000 g for 2 min. The brain supernatant was kept refrigerated in the dark until assayed for dopamine content, which was correlated with the amount of tyrosine hydroxylase, detected by immunoblotting. Light microscopy analysis Brains were removed at set time points and the morphology of cells in the substantia nigra was examined after embedding and staining with methylene blue. Cells were observed for any indications of a reduction in cell size, or the presence of heavily stained condensed nuclei, cytoplasmic and nuclear membrane irregularities, axonal swelling and cell lysis. In addition, since non-apoptotic cell death invokes an inflammatory response, we examined tissues for infiltrating white blood cells. RESULTS Genetic mapping In initial genetic mapping experiments the gene defect in NM3 was localized to mouse chromosome 2 (Figure 1). Subsequent genetic crossing experiments for further fine mapping of this area allowed identification of the genetic locus to within a 1.07-cM distance between D2Mit124 and D2Mit379, which is an area that corresponds to human chromosome 2q24-31. Six hundred and twenty-one mice (121 affected, 500 unaffected) in two different crosses (RIIIS/J x CAST/Ei and RIIIS/J x C57Bl/6J) were utilized to map this gene to its location in an area that does not contain any known genes associated with neurodegeneration or PD.
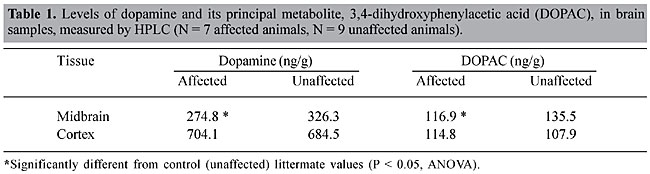
Biochemical analysis of dopamine levels Because of the unique neurological phenotype of NM3, we used HPLC to measure the levels of dopamine, and its principal metabolite 3,4-dihydroxyphenylacetic acid (DOPAC), in both cortex and midbrain samples of affected and unaffected mice (Champney et al., 1992). As predicted, there were decreased neurotransmitter levels in the midbrain of affected animals when compared to control (unaffected) tissue samples (Table 1). The decrease in both dopamine and DOPAC levels corresponds to a 15-20% loss when compared to control tissues.
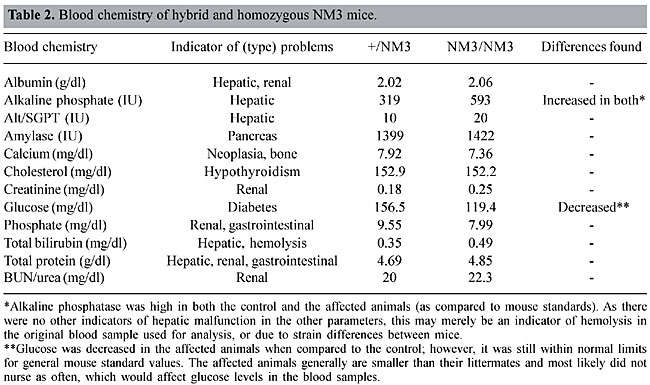
Gross and microscopic analysis No morphological changes were noted in the brain (including the cerebellum, basal ganglia and striatum), inner ear (peripheral vestibular diseases), skeletal muscle (neuromuscular degenerative diseases), peripheral nervous system, or in other organs or organ systems (i.e., hepatic encephalopathies). Clinical pathology Clinical pathologic examination found no problems, ruling out secondary causes of neurological symptoms in the NM3 mouse. Blood/serum chemistries were compared to unaffected littermates in order to control for strain differences among mice, and were within normal limits (Table 2), suggesting no overt pathology in liver, bone, pancreatic, gastrointestinal, endocrine, kidney or hemopoietic systems.
DISCUSSION Positional cloning of disease-associated genes in mice and the discovery of mutations associated with familial PD have not only provided insight into this disease’s mechanisms, but have also enabled the generation of genetically modified animal and in vitro models to dissect specific aspects of the disease. Mutations in three different genes, a-synuclein, PARK2 and DJ-1 have been described in familial PD (Foltynie et al., 2002a,b; Gwinn-Hardy and Farrer, 2002). Other genes associated with parkinsonism include tau (fronto-temporal dementia with parkinsonism; FTDP) and the X-linked recessive syndromes of parkinsonism and dystonia (Arima et al., 1999). a-Synuclein, found in the brains of patients with idiopathic PD, has been identified as a major component of Lewy bodies, suggesting that this substance may be a key player in the pathogenesis of idiopathic PD. Moreover, deletions and point mutations in the parkin gene are associated with autosomal recessive juvenile PD (Shimura et al., 1999), and a single nucleotide polymorphism in the parkin promoter has recently been found to be genetically associated with late-onset idiopathic PD (West et al., 2002). Parkinsonism as a characteristic of tau-associated FTDP suggests that the tau protein may also play a role in the pathogenesis of PD. This is supported by tau immunoreactivity observed in some PD Lewy bodies (Arima et al., 1999) and also by genetic linkage analysis of the tau gene with clinically diagnosed idiopathic PD (Pastor et al., 2000). As tau may play an important role in the axonal transport of vesicles and microtubule-based transport of aggregates, tau could be involved in aggregate formation and/or dopaminergic homeostasis in PD. However, no association between the tau gene and PD cases has been found. It is clear from studies with MPTP that a combination of environmental and genetic research is needed to identify the molecular defects in PD, to elucidate inherited risk factors and ultimately identify the sequence of molecular events that leads to the prototypic pathology of PD. Finding and characterizing genetic models of disease (like the NM3 mouse) now offer hope in our current golden era of neuroscience, in which we strive to address the complex interaction of genetic background and environmental chemical exposure. Ideally, these models should recapitulate the characteristic clinical and pathological features of PD, including hallmark movement disorders and a progressive loss of dopaminergic function. However, this may over-simplify the complexity of PD, and clearly no animal or cell culture model is likely to be an exact phenotypic copy of the human disease. Nonetheless, these models are vital in helping to dissect the many different molecular and biochemical pathways that converge in the pathological phenotype known as PD. We have established the utility of NM3 mice as candidate models for understanding the cellular and molecular pathways underlying the Parkinson’s disease process. Future analysis of the NM3 locus and fine mapping of the genetic region will ultimately be required to identify the defective gene. ACKNOWLEDGMENTS We acknowledge the contributions of Dr. Thomas Champney who help conduct the HPLC analysis. REFERENCES Arima, K., Hirai, S., Sunohara, N., Aoto, K., Izumiyama, Y., Ueda, K., Ikeda, K. and Kawai, M. (1999). Cellular co-localization of phosphorylated tau- and NACP/alpha-synuclein-epitopes in Lewy bodies in sporadic Parkinson’s disease and in dementia with Lewy bodies. Brain Res. 843: 53-61. Albin, R.L. and Greenamyre, J.T. (1992). Alternative excitotoxic hypotheses. Neurology 42: 733-738. Beal, M.F., Hyman, B.T. and Koroshetz, W. (1993). Do defects in mitochondrial energy metabolism underlie the pathology of neurodegenerative diseases? Trends Neurosci. 16: 125-131. Bezard, E., Gross, C.E., Fournier, M.C., Dovero, S., Bloch, B. and Jaber, M. (1999). Absence of MPTP-induced neuronal death in mice lacking the dopamine transporter. Exp. Neurol. 155: 268-273. Carlsson, M. and Carlsson, A. (1990). Interactions between glutamatergic and monoaminergic systems within the basal ganglia - implications for schizophrenia and Parkinson’s disease. Trends Neurosci. 13: 272-276. Champney, T.H., Hanneman, W.H. and Nichols, M.A. (1992). Gamma-aminobutyric acid, catecholamine and indoleamine determinations from the same brain region by high-performance liquid chromatography with electrochemical detection. J. Chromatogr. Biomed. 579: 334-339. Fahn, S. and Cohen, G. (1992). The oxidant stress hypothesis in Parkinson’s disease: evidence supporting it. Ann. Neurol. 32: 804-812. Foltynie, T., Brayne, C. and Barker, R.A. (2002a). The heterogeneity of idiopathic Parkinson’s disease. J. Neurol. 249: 138-145. Foltynie, T., Sawcer, S., Brayne, C. and Barker, R.A. (2002b). The genetic basis of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 73: 363-370. Gwinn-Hardy, K. and Farrer, M. (2002). Parkinson’s genetics: an embarrassment of riches. Ann. Neurol. 51: 7-8. Hallman, H., Lange, J., Olson, L., Stromberg, I. and Jonsson, G. (1985). Neurochemical and histochemical characterization of neurotoxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine on brain catecholamine neurones in the mouse. J. Neurochem. 44: 117-127. Hamre, K., Tharp, R., Poon, K., Xiong, X. and Smeyne, R.J. (1999). Differential strain susceptibility following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration acts in an autosomal dominant fashion: quantitative analysis in seven strains of Mus musculus. Brain Res. 828: 91-103. Langston, J.W. and Irwin, I. (1986). MPTP: current concepts and controversies. Clin. Neuropharmacol. 9: 484-507. Pastor, P., Ezquerra, M., Munoz, E., Marti, M.J., Blesa, R., Tolosa, E. and Oliva, R. (2000). Significant association between the tau gene A0/A0 genotype and Parkinson’s disease. Ann. Neurol. 47: 242-245. Piccoli, F., La Bella, V. and Guarneri, R. (1992). New insights on the pathogenesis of neurodegenerative disorders. Acta Neurol. 14: 455-468. Rathke-Hartlieb, S., Kahle, P.J., Neumann, M., Ozmen, L., Haid, S., Okochi, M., Haass, C. and Schulz, J.B. (2001). Sensitivity to MPTP is not increased in parkinsons disease-associated mutant alpha-synuclein transgenic mice. J. Neurochem. 77: 1181-1184. Shimura, H., Hattori, N., Kubo, S., Yoshikawa, M., Kitada, T., Matsumine, H., Asakawa, S., Minoshima, S., Yamamura, Y., Shimizu, N. and Mizuno, Y. (1999). Immunohistochemical and subcellular localization of Parkin protein: absence of protein in autosomal recessive juvenile parkinsonism patients. Ann. Neurol. 45: 668-672. Tanner, C.M. (1994). Epidemiological clues to the cause of Parkinson’s disease. In: Movement Disorders 3 (Fahn, S. and Marsden, C.D., eds.). Butterworth-Heinemann, Oxford, USA, pp. 124-146. West, A.B., Maraganore, D., Crook, J., Lesnick, T., Lockhart, P.J., Wilkes, K.M., Kapato, G., Hardy, J.A. and Farrer, M.J. (2002). Functional association of the parkin gene promoter with idiopathic Parkinson’s disease. Hum. Mol. Genet. 11: 2787-2792. Willis, G.l. and Donnan, G.A. (1987). Histochemical, biochemical and behavioural consequences of MPTP treatment in C-57 black mice. Brain Res. 402: 269-274. |
|