![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
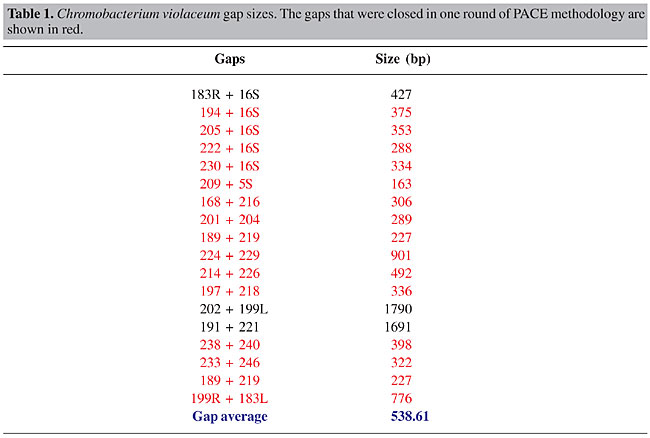
ABSTRACT. In the finishing phase of the Chromobacterium violaceum genome project, the shotgun sequences were assembled into 57 contigs that were then organized into 19 scaffolds, using the information from shotgun and cosmid clones. Among the 38 ends resulting from the 19 scaffolds, 10 ended with sequences corresponding to rRNA genes (seven ended with the 5S rRNA gene and three ended with the 16S rRNA gene). The 28 non-ribosomal ends were extended using the PCR-assisted contig extension (PACE) methodology, which immediately closed 15 real gaps. We then applied PACE to the 16S rRNA gene containing ends, resulting in eight different sequences that were correctly assembled within the C. violaceum genome by combinatory PCR strategy, with primers derived from the non-repetitive genomic region flanking the 16S and 5S rRNA gene. An oriented combinatory PCR was used to correctly position the two versions (copy A and copy B, which differ by the presence or absence of a 100-bp insert); it revealed six copies corresponding to copy A, and two to copy B. We estimate that the use of PACE, followed by combinatory PCR, accelerated the finishing phase of the C. violaceum genome project by at least 40%. Key words: Bacterial genome, PACE, Finishing phase INTRODUCTION Bacterial genome projects usually comprise three well-defined stages. The first one is the construction of high quality random shotgun libraries with varying insert sizes. Following this, the next task is typically to generate a deep coverage of the genome, with random shotgun sequences. This step generates the bulk of the sequences that are used to compile the finished genome sequence, and it is in many ways the heart of the project. Shotgun sequences are then assembled into sequence contigs, each representing a separate, non-overlapping portion of the genome. The resulting assembly is then converted into a continuous genome sequence through a more laborious and time-consuming finishing phase. Due to the small size and low complexity of bacterial genomes, a genome sequence is only considered finished when all bases meet an acceptable quality level and the individual sequences are assembled into a single continuous consensus sequence. During the finishing phase of a bacterial genome project, the overall sequence quality is improved and sequence data are generated to close gaps between existing contigs. Finishing can take a variety of forms, but it almost always involves the generation of sequences from large insert clones that have been demonstrated to span virtual gaps in the shotgun assembly, and by the use of alternative strategies to close real gaps when the corresponding DNA fragment has not appeared in any of the libraries generated for the sequencing project. The occurrence of real gaps is often associated with statistical cloning fluctuation of the shotgun model, repetitive regions in the genome (for example ribosomal operons), and with sequences refractory to the cloning system used. Strategies for real gap closure that have been used so far include the use of alternative cloning vectors, combinatory PCR, with primers derived from contig ends, subtractive hybridization (Frohme et al., 2001), physical mapping (Weinel et al., 2001), and direct sequencing of genomic DNA. The time required for finishing depends on the nature of the genome and most particularly on the structure and frequency of repetitive sequences, but this phase easily outlasts that of the generation of the shotgun sequences and is indeed one of the key rate-limiting steps in bacterial genome sequencing. Here we describe the assembly process and finishing phase of the Chromobacterium violaceum genome project. C. violaceum is a free-living, gram-negative bacteria that is highly abundant in the water and banks of the Negro River in the Brazilian Amazon, and produces a violacein pigment (Caldas, 1990; Duran and Menck, 2001) with antimicrobial activity against some important pathogens (Duran et al., 1994; De Souza et al., 1998; Leon et al., 2001) as well as antiviral (Duran and Menck, 2001) and anticancer (Melo et al., 2000) activity. The sequencing and analysis of the C. violaceum genome were entirely executed by the Brazilian National Genome Sequencing Consortium. A random shotgun strategy was used, and the resulting sequences were assembled into 57 contigs. These were then organized into 19 scaffolds, using the information from shotgun and cosmid clones; sequences of which were located in different contigs. Forty-seven virtual gaps within the scaffolds were closed by whole insert sequencing of the corresponding shotgun/cosmid clones. Eighteen real gaps were, for the most part, closed by applying the PCR-assisted contig extension (PACE), followed by specific confirmatory and oriented combinatory PCR. PACE involves the generation of stepwise extensions from the ends of contigs by PCR, until the closure of individual gaps is achieved. This methodology has proven to be especially useful for extending the multicopy ribosomal operons, and has greatly accelerated the finishing phase of the C. violaceum genome project. METHODS PACE methodology PACE was developed as a two-step PCR strategy, with nested primers derived from contig ends, as described by Carraro et al., 2003. Briefly, a set of 96 primers was randomly selected, with no reference to their precise sequence. Pairs of outward-facing specific nested primers were then designed, approximately 140 bp from contig ends and 40 bp apart from each other. Specific primers were checked for alignment to a single position of the genome with the FASTA program. Secondary structures and dimer formation were verified using the Oligo Tech software (http://www.oligosetc.com/analysis.php). PCR was then performed in 96-well plates, using 80 ng genomic DNA as templates for the first reaction and 1 µl of a 1:100 dilution of the first reaction as templates in a subsequent nested reaction (Figure 1). 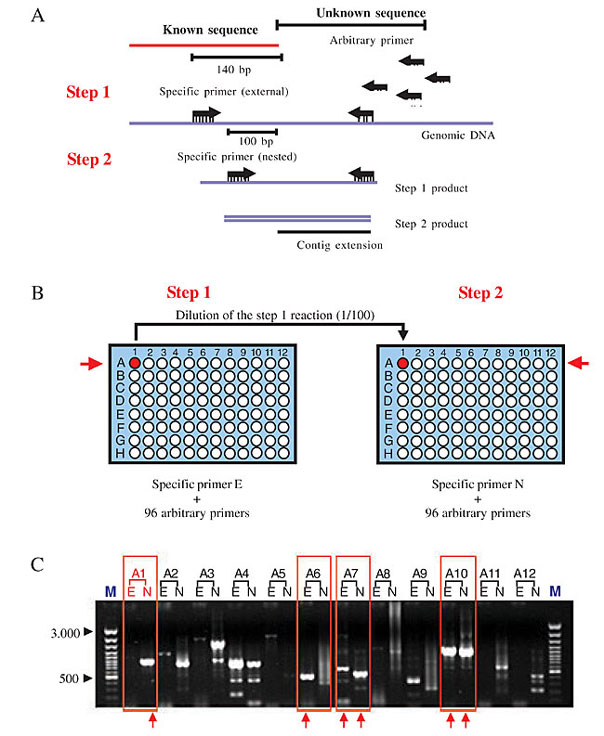
Figure 1. A schematic outline of the PACE reaction. A, The PACE strategy with the schematic position of specific external and nested primer, and the arbitrary primers in the step 1 and step 2 reactions. B, Representation of step 1 and step 2 PCR reactions. Arbitrary primers (0.5 mM) were distributed with an 8-channel pipette in an ordered manner on the 96-well plate. In step 2, 2 µl of a 1:100 dilution of the step 1 reaction products was transferred to a second 96-well plate, with the same 8-channel pipette and 0.5 mM of the nested specific primer and 0.5 mM of the same arbitrary primer used in step 1 used as a template. C, Three microliters of each PCR reaction was loaded into adjacent lanes onto a 1% ethidium bromide-stained agarose gel and those with single detectable bands were selected for purification and sequencing (red box). E = external and N = nested PACE products.
Specific PCR All contig extensions were confirmed by specific PCR, followed by direct sequencing of the resulting PCR products. Reaction mixtures for specific PCR contained 80 ng genomic DNA, 250 µM dNTP, 1.5 mM MgCl2, 1 U Platinum High Fidelity Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA), and 10 µM of each specific primer in a final volume of 20 µl. Reactions were carried out at 94°C for 30 s, 60°C for 30 s, and 68°C for 2 min from 35 cycles. An initial cycle of 94°C for 2 min, and a final extension at 68°C for 6 min was used. Combinatory PCR For ribosomal operon orientation, primers were designed in the flanking regions of the 16S and 5S rRNA genes (outside of the operon repeat unit) and PCRs were undertaken in a combinatory way. The expected fragment sizes were between 5.2 and 5.8 kb, depending on the annealing position of the primers in the flanking region. PCRs were performed in a final volume of 20 µl, containing 1.5 mM MgCl2, 300 µM dNTPs, 2 U Platinum High Fidelity Taq DNA polymerase (Invitrogen) and 15 µM of each flanking primer. Reactions were carried out at 94°C for 30 s, 60°C for 30 s and 68°C for 5 min, for 35 cycles. Initial denaturation step at 94°C for 2 min, and final extension step at 68°C for 6 min, was used. Two almost identical versions of the rRNA operon were identified during the assembly phase. The difference between the two copies resides in a 100-bp insertion in the intergenic region between the 16S and ILE genes and a 74-bp insertion between the ILE and ALA genes (copy A - contains 100 bp-16S/ILE, 74 bp-ILE/ALA and copy B - does not contain 100 bp-16S/ILE, 74 bp-ILE/ALA). In order to position correctly the two versions in the genome, a primer located 150 bp downstream of the 100-bp insertion (in the ILE gene) was used in a combinatory way in amplification reactions, together with one of the eight specific primers corresponding to the 16S rRNA flanking region. PCR reactions were carried out as described above, except for modifications in the cycling parameters (94°C for 30 s, 60°C for 30 s and 68°C for 2 min). Sequence specificity was checked by BLASTN analysis of the two fragment extremities against the two available ribosomal operon copies. A fragment was considered specific to one of the two copies if the high quality sequence portion aligned with the corresponding copy with at least 95% identity. Product analysis Three to five microliters from each PCR were loaded onto a 1% ethidium bromide-stained agarose gel. Single PACE products, or fragments of expected size in the case of confirmatory or combinatory PCRs, were purified with the QIAquickTM PCR Purification Kit (Qiagen, Valencia, CA, USA) and sequenced directly using the same specific primer used for amplification on an ABI PrismR 3100 DNA sequencer (Applied Biosystems, Foster City, CA, USA). High-quality sequences with more than 300 bp, and Phred (Gordon et al., 1998) quality greater than 20, were analyzed for specificity. Sequence specificity was checked by BLASTN analysis against the available genome assembly, and a fragment was considered specific if at least 25 bp of the high quality sequence aligned with at least 95% identity with the end of the corresponding contig. RESULTS Chromobacterium violaceum genome assembly The sequencing and analysis of the C. violaceum genome were entirely executed by the Brazilian National Genome Sequencing Consortium, comprising 25 sequencing laboratories, one bioinformatics center, and three coordination laboratories, distributed throughout Brazil. In the initial phase of the project, random shotgun sequencing produced approximately 80,000 high quality reads (with PHRED score >20), generated from both ends of pUC18 clones, with insert sizes raging from 2.0 to 4.0 kb. Additionally, both ends of 3,350 cosmid clones, with an average insert size of 40 kb, were also sequenced, providing a validation check of the final assembly. The shotgun sequences, corresponding to approximately 13-fold genome coverage, were assembled into 57 contigs. These shotgun contigs were then organized into 19 scaffolds, using the information from shotgun and cosmid clones, the end sequences of which were located in different contigs. Forty-seven virtual gaps within the 19 scaffolds were closed by whole insert sequencing of the corresponding shotgun/cosmid clones. Points of genome assembly instability and regions of low quality sequences were identified using the Autofinisher Program, and were resolved by re-sequencing of the selected clones. Real gaps that did not involve the rRNA gene (n = 18) were mainly closed by applying PACE, as detailed in Carraro et al. (2003). Here, we relate the methodology used to close rRNA-related gaps and to correctly position the eight ribosomal operon units in the C. violaceum genome assembly. Closure of real gaps by PACE The PACE technique depends on rare mismatched primer-template interactions that occur between arbitrary primers and template DNA with an unknown sequence, even under highly stringent conditions. These are captured through elevated PCR-cycle repetition, and through the use of specific anchoring primers, corresponding to adjacent regions of known sequence (contig ends). Thus, PACE allows the generation of stepwise extensions from the ends of contigs by PCR, until the closure of individual gaps is achieved. When we started using PACE to close the C. violaceum genome assembly, seven of the existing 38 contig ends ended with the 5S rRNA sequence and three ended with the 16S rRNA sequence, suggesting the existence of at least seven identical copies of the rRNA operon (Figure 2). Twenty-two PACE reactions (Figure 2) were initially applied to extend contig ends that did not contain rRNA-derived sequences, resulting in 137 specific sequences, allowing the immediate closure of 15 real gaps in the C. violaceum assembly due to their relatively small size (Table 1). Of these, six apparently linked the contig to an rRNA gene (one to a 5S rRNA gene and five to 16S rRNA genes), suggesting a total of eight copies of the ribosomal operon. In two cases (gap 191-221 of 1691 bp, gap 202-199L of 1790 bp), additional rounds of PACE were undertaken until the complete closure of each gap was achieved. All contig extensions and gap closures were confirmed by specific PCR, followed by direct sequencing of the PCR products. 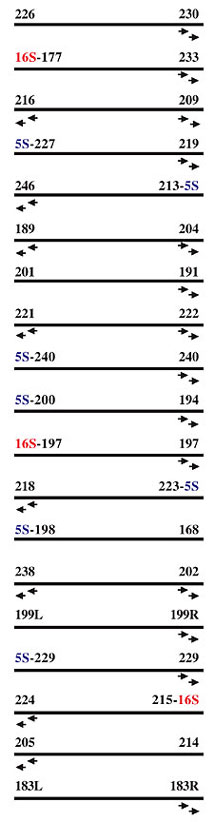
Figure 2. Representation of the scaffold result of the Chromobacterium violaceum genome assembly - 16S and 5S - rRNA genes. The black arrows indicate the C. violaceum contig extremities where the pair of external and nested primers (E and N) was derived.
Closure of gaps related to rRNA sequences We used a single pair of nested primers specific for the 16S rRNA gene to check for the extension of contigs ending in 16S rRNA. Thirty-five PACE fragments were selected for sequencing and high quality sequences were further analyzed using BLASTN to check for their specificity. Of these, 34 were found to be specific, and they confirmed five novel flanking regions for the 16S rRNA gene (contigs 194, 205, 222, 230 and 183), in addition to the three known (contigs 177, 197 and 215), giving a total of eight rRNA operons (Figure 3). 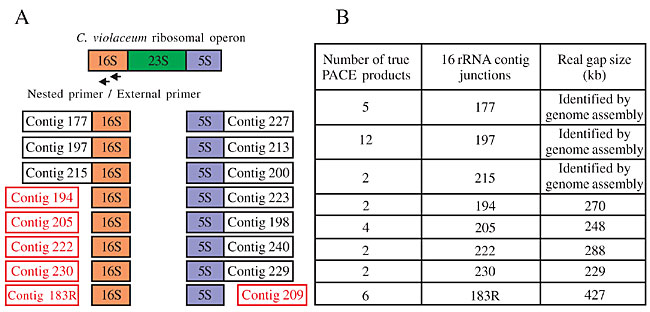
Figure 3. Representation of the Chromobacterium violaceum ribosomal operon. A, The seven 5S rRNA junctions revealed by the initial genome assembly and the three 16S rRNA junctions. The red box shows the six junctions identified by PACE methodology. B, The table shows the number of PACE products applied in the 16S rRNA gene for each contig extension (columns 1 and 2) and the size of the real gaps (column 3).
All 16S and 5S rRNA contig extensions were confirmed by specific PCR, followed by direct sequencing of the PCR products. A schematic outline of the final C. violaceum genome assembly is presented in Figure 4A. Seventy-two specific PCR reactions, including real gaps and rRNA-related gaps, were performed, corresponding to the four possible combinations between the external and nested primers from both contig ends (Figure 4B). At least one fragment from each junction was submitted for sequencing to check the specificity. 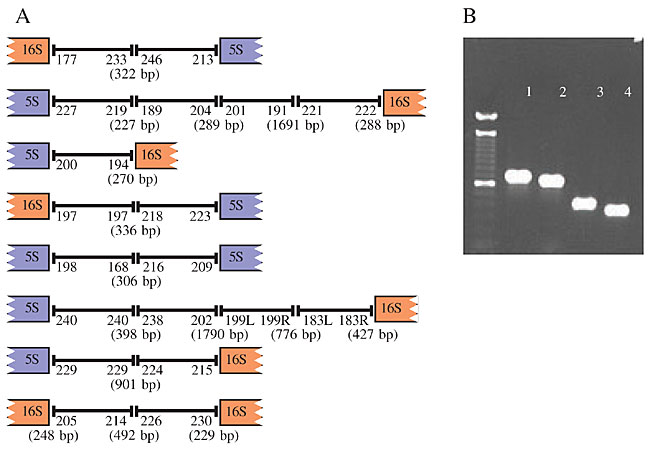
Figure 4. A, A schematic outline of the final genome assembly of Chromobacterium violaceum achieved by PACE methodology, showing the contig ends and the sizes of the gaps. B, Confirmatory PCR using a combinatory format with nested and external primers of contig ends 189 and 219 - (1) primers 189 E + 219 E; (2) 189 E + 219 N; (3) 189 N + 219 E, and (4) 189 N + 219 N - (E - external primer and N - nested primer).
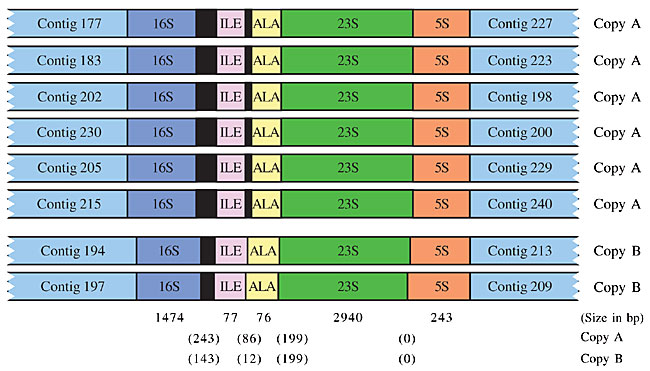
Positioning of the eight ribosomal operons in the Chromobacterium violaceum genome assembly After the 16S PACE protocol, the genome assembly was composed of eight unoriented contigs, ending either with 16S rRNA or 5S rRNA sequences. To assemble the contigs in the correct order, we used a combinatory PCR strategy, with primers derived from the non-repetitive genomic region flanking the 16S and 5S rRNA gene. PCR fragments of expected size were sequenced from both extremities, and high quality sequences were aligned to the genome assembly. The orientation of the contigs was confirmed if at least 100 bp of the sequenced fragment aligned with the end of the corresponding contigs, outside the common ribosomal operon sequence (Figure 5).
Figure 5. The oriented eight Chromobacterium violaceum ribosomal operons (16S-ILE-ALA-23S-5S) related to the 16S and 5S rRNA-flanking contigs. The intergenic region (in parentheses) and gene size are shown. The two ribosomal operon versions are represented, and the differences in the intergenic regions are shown. The whole genome assembly revealed a slight difference between the sequences, corresponding to the ribosomal operons. Two almost identical versions were identified. The difference between the two versions resided in a 100-bp insertion between the 16S and ILE genes, and 74 bp between the ILE and ALA genes. In order to correctly place the two versions (A and B), in the eight already-oriented ribosomal operon copies, a primer located 150 bp downstream of the 100-bp insertion was used in a combinatory way, together with one of the eight specific primers corresponding to the 16S rRNA-flanking region. Using this strategy, we found out that most (6/8) of the rRNA operon copies contained the 100-bp and 74-bp insertion (copy A). These results were independently confirmed by the finding of a larger number of reads in the genome assembly in which the 100-bp insertion was not present. DISCUSSION After the initial assembly of shotgun reads, the C. violaceum genome sequence was organized into 19 scaffolds. The genome sequence assembly was mainly made difficult by the eight almost identical rRNA operon copies dispersed throughout the genome. Using PACE, we were able to generate contig extensions with an average of 1 kb in length from all contigs, which closed the majority of gaps in a single round of experimentation. In addition, the PACE methodology proved to be extremely useful for extending the multicopy ribosomal operons. The surprisingly high success rate of our approach can be attributed to deep shotgun coverage and the small size of the gaps in the C. violaceum genome assembly. However, deep shotgun coverage is not a pre-requisite for using PACE. Analyses made by our group demonstrated that the methodology can be applied at early stages of the bacterial genome assembly, drastically reducing the time and cost of the finishing phase. Usually the finishing phase of a genome project takes between 50 to 60% of the total time of the project (Simpson, 2001). In the case of the C. violaceum genome project we used approximately 30% of the total time required to conclude the project, corresponding to a reduction of at least 40% of the estimated time. The importance of PACE is that, at a pre-determined point in the shotgun sequencing, primers can be generated and contig extensions obtained with no requirement for a one-by-one analysis of the gaps. Using successive PACE reactions, it is possible to close gaps in bacterial genomes in a stepwise fashion, regardless of their size. Based on the experience accumulated in the C. violaceum genome project we strongly recommend the use of the PACE methodology in the finishing phase of bacterial genome projects. ACKNOWLEDGMENTS The work described here was undertaken within the context of the Brazilian National Genome Program (a consortium funded in December 2000 by the Ministério da Ciência e Tecnologia - MCT - through the Conselho Nacional de Desenvolvimento Científico). We thank André Vettore for helping in the corrected positioning of ribosomal operon in the genome assembly and Eduardo Abrantes for helping in the preparation of the figures for this manuscript. REFERENCES Caldas, L.R. (1990). Um pigmento nas águas negras. Cienc. Hoje 11: 55-57. Carraro, D.M., Camargo, A.A., Salim, A.C.M., Grivet, M., Vasconcelos, A.T. and Simpson, A.J.G. (2003). PCR-assisted contig extension: stepwise strategy for bacterial genome closure. Biotechniques 34: 626-632. De Souza, A.O., Aily, D.C., Sato, D.N. and Duran, N. (1998). In vitro activity of N,N-dimethyl-2-propen-1-amines against Mycobacterium tuberculosis. J. Antimicrob. Chemother. 42: 407-408. Duran, N. and Menck, C.F. (2001). Chromobacterium violaceum: a review of pharmacological and industrial perspectives. Crit. Rev. Microbiol. 27: 210-222. Duran, N., Antonio, R.V., Haun, M. and Pilli, R.A. (1994). Biosynthesis of a Trypanocide by Chromobacterium violaceum. World J. Microbiol. Biotechnol. 10: 686-690. Frohme, M., Camargo, A.A., Czink, C., Matsukuma, A.Y., Simpson, A.J.G. and Verjovski-Almeida, S. (2001). Direct gap closure in large-scale sequencing project. Genome Res. 11: 901-903. Gordon, D., Abajian, C. and Green, P. (1998). Consed: a graphical tool for sequence finishing. Genome Res. 8: 195-202. Leon, L.L., Miranda, C.C., De Souza, A.O. and Duran, N. (2001). Antileishmanial activity of the violacein extracted from Chromobacterium violaceum. J. Antimicrob. Chemother. 48: 449-450. Melo, P.S., Maria, S.S., Vidal, B.C., Haun, M. and Duran, N. (2000). Violacein cytotoxicity and induction of apoptosis in V79 cells. In Vitro Cell Dev. Biol. Anim. 36: 539-543. Simpson, A.J.G. (2001). Genome sequencing networks. Nat. Rev. 2: 979-983. Weinel, C., Tummler, H., Hilbert, H., Nelson, K.E. and Kiewitz, C. (2001). General method of rapid Smith/Birnstiel mapping adds for gap closure in shotgun microbial genome sequencing projects: application to Pseudomonas putida KT2440. Nucleic Acids Res. 29: E110. |
|