
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
ABSTRACT. A fluid genome is a great advantage to prokaryotes, enabling quick adaptation to various types of ecological niches and to diverse environmental selective pressures. A substantial portion of these sudden changes is mediated by lateral gene transfer (LGT), through genetic recombination mechanisms, such as transformation, conjugation and transduction. The recent sequencing of several organisms has offered a new approach to the study of LGT, using comparison and analysis of nucleotide sequences dispersed throughout the genome of these species. This analysis in Choromobacterium violaceum has revealed four prophage and 12 insertion sequences, suggesting genetic exchange with several other bacterial species, including Salmonella enterica, Ralstonia and Xanthomonas. An Rhs (recombination hot spot) element (containing a vgr-like gene) was also observed, the function of which remains unknown, but it has a sequence related to species of Acinetobacter and Sphingomonas. These results support the role of LGT in the acquisition of new traits by C. violaceum. Key words: Bacteriophages, Transposons, Rhs elements, Chromobacterium violaceum INTRODUCTION The genome of more than 100 species of bacteria has been sequenced, and each day more sequence data are generated. The comparison and the organization of these different genomes reveal interesting biological and evolutionary information. In fact, the genomes of bacteria are remarkably fluid (Boucher et al., 2003). A substantial portion of the bacterial genome has not been inherited from the parental cells; rather it has been acquired horizontally by a lateral gene transfer (LGT) process, mediated by genetic recombination mechanisms, such as transformation, conjugation and transduction (Doolittle, 1999; Boucher et al., 2003). Several lines of evidence indicate that LGT has played a major role in bacterial evolution, disseminating key traits both within and among bacterial species (Boucher et al., 2003). The acquisition of new traits by this means could allow a sudden adaptation of bacteria to new environment conditions. Acquisition of new phenotypes by LGT is considered a “quantum leap” in evolution, as the genes generally are transferred laterally en bloc, forming “islands” of heterologous DNA in the new host. Considering the fluidity of bacterial genomes, the exchange of DNA by transduction, a process mediated by bacteriophages (or simply phages), seems to play an essential role (Canchaya et al., 2003a,b; Casjens, 2003). In fact, in several cases, it is impossible to understand the biology of bacteria without understanding the biology of their phages. Bacteriophages are extremely varied and their diversity remains uncharacterized (Brüssow and Hendrix, 2002; Rohwer, 2003). Different types of phage virions may carry single- or double-stranded DNA or RNA, exhibit different morphologies, and use different strategies for replication. Ninety-six percent of all bacterial viruses are tailed phages (Caudovirales), which are classified as Myoviridae, Siphoviridae and Podoviridae, based on the type of tail present (Brüssow and Hendrix, 2002). Some phages are only able to trigger a lytic replication cycle. Others, however, the so-called temperate phages, can “choose” between a lytic or a lysogenic state. Thus, infection of a bacterial cell with a temperate phage can have two outcomes: multiplication of the virion with lysis of bacteria, or lysogenization, where the DNA of the virus is integrated into the bacterial chromosome (Campbell, 2001). When this latter event takes place, the bacteria can acquire new traits. In fact, it is well established that integrated virus genomes (prophages) are responsible for conferring to the bacterial host the ability to produce a variety of exotoxins, colonization factors, bacteriocins, resistance to serum and phagocytes, and other traits (Nakayama et al., 2000; Boyd and Brüssow, 2002; Wagner and Waldor, 2002; Davis and Waldor, 2003). Most of the genomes of bacteria published to date carry prophages, and it is not rare for bacteria to contain multiple prophages (Canchaya et al., 2003b; Casjens, 2003). An extreme example is represented by the food-borne pathogen E. coli O157:H7 strain Sakai, which contains 18 prophages that account for 16% of its genome content (Hayashi et al., 2001). Several unique DNA sequences of the Sakai strain, including genes coding for pathogenic factors are present in the genome of its phages (Hayashi et al., 2001). The same observation is valid for other bacterial species (Figueroa-Bossi et al., 2001; Boyd and Brüssow, 2002; Wagner and Waldor, 2002; Boucher et al., 2003; Canchaya et al., 2003a; Casjens, 2003; Porwollik and McClelland, 2003). Insertion sequences (IS) and transposons are present in virtually all prokaryotes. These mobile elements are also implicated in the transference of new traits between different strains or species of bacteria (Craig et al., 2002). The completion of genomic sequencing of C. violaceum ATCC 12472 (Vasconcelos et al., 2003) has revealed the presence of four prophages, and some open reading frames (ORFs) with similarity to phage genes, dispersed in the genome. There are also 12 transposase genes; two of them are part of an Rhs (recombination hot spot) element. These mobile elements could have a role in recombination processes and gain of function by C. violaceum. RESULTS AND DISCUSSION Bacteriophages The positions of the four prophages and the Rhs element in the genome of C. violaceum were determined (Figure 1). The phages were named CvP1 to CvP4, abbreviations for C. violaceum phages 1 to 4. The position of the Rhs element is also indicated. 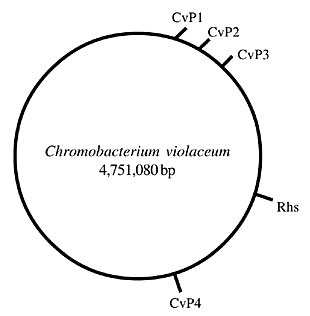
Figure 1. Position of the CvP prophages and the Rhs (recombination hot spot) element in the Chromobacterium violaceum genome. CvP1-CvP4 = C. violaceum phages 1 to 4.
CvP1 Bacteriophage CvP1 is composed of at least 20 ORFs (CV0337 to CV0356), accounting for 19436 bp. The GC ratio is 66.3%, which is quite close to the 64.83% of the C. violaceum genome. The blast analysis of its ORFs indicated that CvP1 is a Mu-like phage. Various ORFs are similar to those of phage FluMu of Haemophilus influenzae and to Neisseria meningitidis Mu-like prophages (Morgan et al., 2002). Bacteriophage Mu is a double-strand DNA phage, with a wide host range and distinctive properties when compared with other phages (Morgan et al., 2002). Mu prophage formation is accompanied by integration into nearly random chromosomal locations that generally lead to detectable mutations in the host. Additionally, Mu replicates by transposition, unlike other known types of phages (Campbel, 2001). An ORF with similarity to a DNA transposition gene of Neisseria meningitidis was found in the CvP1 genome (CV0355). The CvP1 genome is smaller than that of Mu and other Mu-like phages, such as FluMu of H. influenzae and Pnm1 of Neisseria meningitides, suggesting that it is defective. CvP2 The CvP2 bacteriophage is composed of at least 27 ORFs (CV0406 to CV0432), accounting for a region of 22408 bp. The GC ratio (63.73%) is very similar to the GC content of the C. violaceum genome. Most of the CvP2 ORFs exhibit similarities to tail and fiber proteins of phages found in Salmonella enterica subsp. enterica and in Pseudomonas sp. Regulatory genes are also present, as a phage Mu middle regulator (CV0406) and a late gene expression regulator (CV0416). Interestingly, there are ORFs with similarity to Carotovoricin Er of Erwinia carotovora and the R-type pyocins of Pseudomonas sp. Carotovoricin Er is a phage-tail-like bacteriocin produced by E. carotovora subsp. carotovora strain Er, a causative agent of soft rot disease in plants (Nguyen et al., 2001). The R-type pyocins resemble non-flexible and contractile tails of bacteriophages. They provoke a depolarization of the cytoplasmic membrane, with pore formation (Michel-Briand and Baysse, 2002). These observations suggest that CvP2 has bacteriocin activity. In fact, it has been found that lysates of C. violaceum strain ATCC 12472 inhibit the growth of a wide range of Gram-positive and Gram-negative bacteria, including Staphylococcus aureus, Streptococcus sp., Bacillus cereus, Pseudomonas aeruginosa, E. coli, Salmonella enterica (Ackermann and Gauvreau, 1972). Some CvP2 ORFs are also related to phage P2 and Mu, indicating its genetic complexity. CvP3 The CvP3 is composed of few ORFs (CV0645 to CV0652), identified as being of phage origin because they exhibit similarity to genes of P2 and P4 phages of E. coli. The GC content is lower (58.97%) than that of the C. violaceum genome, indicating a heterologous origin. Two hypothetical ORFs (CV0649 and CV0650) are located between a regulatory and an integrase gene, and they have lower GC percentages, 47.6 and 35.61%, reinforcing the heterologous origin of this element. It is also flanked by a number of hypothetical ORFs that could belong to the CvP3 genome. CvP4 The CvP4 is composed of about 51 ORFs (CV2114 to CV2150), corresponding to a region of 27668 bp. CvP4 seems to be a chimeric prophage, containing ORFs with similarity to different phages. The tail and tail fiber genes are similar to P2-like phage genes (Rishovd et al., 1998). These genes exhibit similarity to a P2-like prophage of S. enterica subsp. enterica serovar Typhi, the so-called CT18 phages (Parkhill et al., 2001; Canchaya et al., 2003b). Other ORFs exhibit similarity to other phage genes, such as Mu and phages of Erwinia carotovora. Interestingly, there are a number of conserved hypothetical ORFs with similarity to genes of S. enterica subsp. enterica Typhi. We can speculate that CvP4 was responsible for introducing a number of genes from S. enterica to the C. violaceum genome. The presence of a gene (CV2157), with motifs of a type II secretion protein of V. cholerae, is also interesting. A gene with these characteristics was also found in the genome of CT18-02 (Canchaya et al., 2003b), reinforcing the similarity between CvP4 and the CT18 phages. Reminiscent phages There are two ORFs (CV3545 and CV3546) with similarity to hypothetical phage ORFs of Yersinia pestis (Deng et al., 2002). However, the genes flanking these ORFs do not exhibit similarity to phage genes. In Y. pestis, those ORFs are localized between toxin genes, and they do not seem to compose a functional phage. The CV3861 is a conserved hypothetical ORF with motifs of a phage-related tail transmembrane protein found in Ralstonia solanacearum (Salanoubat et al., 2002). Insertion sequences There are 12 ORFs coding for transposases and forming IS in the C. violaceum genome. Five of them (CV0004, CV1248, CV1923, CV2224, and CV2556) are homologous to the IS1404 of Xanthomonas campestris pv. campestris (da Silva et al., 2002), with an identity of about 65%. IS1404 belongs to the IS3 family of insertion sequences. These more abundant IS of C. violaceum are almost identical to each other, and they have a GC content (58.4%) different from that of C. violaceum, indicating a heterologous origin. There are two ORFs (CV0957 and CV0958) homologous to ORFb and ORFa of IS1421, respectively. IS1421 is a transposable element member of the IS5 family present in R. solanacearum (Salanoubat et al., 2002). Again, the GC content is lower (60.19% for ORFa and 57.43% for ORFb) than that of the C. violaceum genome. The other ORFs present similarity to the IS found in Ralstonia sp. (CV2316), Sphingomonas sp. (CV3035) and Anabaena (CV3036). Recombination hot spot element A remarkable characteristic of the C. violaceum genome is the presence of a putative Rhs element. The Rhs elements are complex genetic composites, well studied in E. coli (Hill et al., 1994). The prototypical structure of this family of elements is characterized by a GC-rich core region of about 3.7 kb, forming a single-open reading frame, and an adjacent AT-rich core extension. The core ORF is followed or overlapped by a shorter ORF, called the downstream ORF (dsORF), also AT-rich. Another feature common to Rhs elements is the presence of one or more IS, positioned to the right of the dsORF (Hill et al., 1994). An element with a similar structure is present in C. violaceum. It is formed by a longer core region (CV1431), followed by an AT-rich region, probably corresponding to the core extension, and a shorter ORF (CV1428), corresponding to the dsORF. Two IS (CV1426 and CV1427) are associated with this element. Differently from E. coli, they do not exhibit similarity to H-rpt, the most frequently observed IS of E. coli Rhs elements (Zhao et al., 1993). On the contrary, the transposases of the C. violaceum Rhs element exhibit similarity to transposases of Acinetobacter sp. and Sphingomonas sp. Despite the presence of IS, there has been no experimental demonstration of Rhs movement (Hill et al., 1994). However, a possible role of this element in site-specific recombination was proposed, although this recombination event seems to occur much less frequently than has been observed for other mobile elements, such as phages and IS (Hill et al., 1994). Wang et al. (1998) proposed the inclusion of a new ORF called vgr as a structural component of some Rhs elements. This proposal was based on the close linkage of vgr to the core region of the RhsG and RhsE elements of E. coli. However, in other cases the vgr is absent, which argues against the inclusion of this gene as a component of Rhs. Adjacent to the core region of the C. violaceum Rhs element, spaced by only 41 bp, there is a vgr-like ORF. The presence of vgr in Rhs elements found in bacterial species phylogenetically distant from each other, such as E. coli and C. violaceum, led us to believe that this gene must be a structural component of at least some Rhs elements. On the contrary, no homolog to the Vibrio cholerae hemolysin-corregulated protein (hcp) was found, which reinforces the hypothesis of Wang et al. (1998) that this gene is not a component of Rhs, despite its presence in the RhsG of E. coli. The biological function of Rhs elements remains unknown. They are not essential for the cell because there are E. coli isolates that do not contain this element (Hill et al., 1994, 1995). However, they are widespread in natural E. coli populations and their presence correlates with the population structure (Hill et al., 1995), indicating that these elements are maintained by selective pressures. The predicted core protein sequence is similar to the sequence of a Bacillus subtilis wall-associated protein, leading to the speculation that the core product is a cell surface protein and has a binding function (Hill et al., 1994). These authors also hypothesized that this element provides the cell with an advantage in a specific habitat, such as a mammalian host or the soil/water environment. CONCLUDING REMARKS There are papers on phages in C. violaceum (Ackermann and Gauvreau, 1972; Rucinsky et al., 1972; Rucinsky and Cota-Robles, 1973). These studies demonstrated bacteriophage tail-like particles in cultures of C. violaceum after induction with UV light or mitomycin C. The information from the genomic sequencing of this bacterium confirms this pioneering work. The CvP prophages are of different origins, and they have a mosaic genome, a characteristic shared with the majority of the phages sequenced so far. The CvP apparently had an important role in the biology of C. violaceum, probably promoting exchange of genetic material with other species. Interestingly, some prophages exhibit a GC content close to that of the bacterium genome, indicating that they are ancient elements or that they were acquired from organisms with similar percentages of GC. For instance, CvP1 probably was acquired prior to the divergence of C. violaceum and N. meningitidis, as indicated by the similarity between ORFs of CvP1 and Pmn1. The study of these prophages also has a potential to find practical applications. For instance, it is interesting to explore the potential use of CvP2 prophage as a bacteriocin, as revealed by the analysis of its genome and from previous experimental data (Ackermann and Gauvreau, 1972). The presence of IS in the genome also indicates that there has been exchange of genetic material between C. violaceum and other free-living bacteria, such as Xanthomonas sp. and Ralstonia. The IS3 family is strongly represented, particularly by the presence of IS1404. The GC contents of these IS are different from that of the C. violaceum genome, indicating a heterologous origin. A remarkable characteristic of C. violaceum ATCC 12472 is the presence of an Rhs element. Our data reinforce the proposal of Wang et al. (1998), who considered the vgr gene a structural component of at least some Rhs elements. We can only speculate on the biological function of this element, but its presence in C. violaceum suggests a function in the adaptation to a free-living mode of life. It would interesting to investigate if Rhs is widespread in natural populations of C. violaceum. If this is the case, it would be an attractive element to be used in studies of the clonal structure of populations. C. violaceum is a b-proteobacterium phylogenetically related to Neisseria meningitidis and belonging to the Neisseriaceae family. However, the comparison of the ORFs of C. violaceum with those of other organisms reveals a considered close similarity to ORFs of free-living organisms, including R. solanacearum and P. aeruginosa. These ORFs are particularly related to this bacterium’s interaction with the environment. On the other hand, the similarity to N. meningitidis involves ORFs related to housekeeping functions (Vasconcelos et al., 2003). Thus, environmental adaptation is to some extent due to the presence or absence of particular ORFs within the genome. The presence of mobile elements, such as prophages and IS, exhibiting similarity to ORFs of free-living organisms, is indicative of lateral gene transfer between C. violaceum and other free-living organisms and could explain the acquisition of genes that probably performed a central role in the evolution and adaptation of C. violaceum to its present mode of life. ACKNOWLEDGMENTS R. Almeida, P.B. Trevilato, L.A. Bartoleti and J.L. Proença-Módena were supported by grants from the CNPq/RHAE. The C. violaceum genome-sequencing project was supported by funds from the MCT (Ministério da Ciência e Tecnologia) and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) in the Brazilian National Genome Project Consortium. REFERENCES Ackermann, H.W. and Gauvreau, L. (1972). Defective phages in Chromobacterium. Zentralbl. Bakteriol. 221: 196-205. Boucher, Y., Douady, C.J., Papke, R.T., Walsh, D.A., Boudreau, M.E., Nesbo, C.L., Case, R.J. and Doolittle, W.F. (2003). Lateral gene transfer and the origins of prokaryotic groups. Annu. Rev. Genet. 37: 283-328. Boyd, E.F. and Brüssow, H. (2002). Common themes among bacteriophage-encoded virulence factors and diversity among the bacteriophages involved. Trends Microbiol. 10: 521-529. Brüssow, H. and Hendrix, R.W. (2002). Phage genomics: small is beautiful. Cell 108: 13-16. Campbell, A.M. (2001). Bacteriophages. In: Fields Virology (Knipe, D.M. and Howley, P.M., eds.). 4th edn. Lippincott Willians & Wilkins, Philadelphia, PA, USA, pp. 659-682. Canchaya, C., Fournou, G., Chibani-Chennoufi, S., Dillmann, M.L. and Brussow, H. (2003a). Phage as agents of lateral gene transfer. Curr. Opin. Microbiol. 6: 417-424. Canchaya, C., Proux, C., Fournous, G., Bruttin, A. and Brussow, H. (2003b). Prophage genomics. Microbiol. Mol. Biol. Rev. 67: 238-276. Casjens, S. (2003). Prophages and bacterial genomics: what have we learned so far? Mol. Microbiol. 49: 277-300. Craig, N.L., Craigie, R., Gellert, M. and Lambowitz, A. (2002). Mobile DNA II. American Society for Microbiology, New York, NY, USA. da Silva, A.C., Ferro, J.A., Reinach, F.C., Farah, C.S., Furlan, L.R., Quaggio, R.B., Monteiro-Vitorello, C.B., Van Sluys, M.A., Almeida, N.F., Alves, L.M., do Amaral, A.M., Bertolini, M.C., Camargo, L.E., Camarotte, G., Cannavan, F., Cardozo, J., Chambergo, F., Ciapina, L.P., Cicarelli, R.M., Coutinho, L.L., Cursino-Santos, J.R., El-Dorry, H., Faria, J.B., Ferreira, A.J., Ferreira, R.C., Ferro, M.I., Formighieri, E.F., Franco, M.C., Greggio, C.C., Gruber, A., Katsuyama, A.M., Kishi, L.T., Leite, R.P., Lemos, E.G., Lemos, M.V., Locali, E.C., Machado, M.A., Madeira, A.M., Martinez-Rossi, N.M., Martins, E.C., Meidanis, J., Menck, C.F., Miyaki, C.Y., Moon, D.H., Moreira, L.M., Novo, M.T., Okura, V.K., Oliveira, M.C., Oliveira, V.R., Pereira, H.A., Rossi, A., Sena, J.A., Silva, C., de Souza, R.F., Spinola, L.A., Takita, M.A., Tamura, R.E., Teixeira, E.C., Tezza, R.I., Trindade dos Santos, M., Truffi, D., Tsai, S.M., White, F.F., Setubal, J.C. and Kitajima, J.P. (2002). Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature 417: 459-463. Davis, B.M. and Waldor, M.K. (2003). Filamentous phages linked to virulence of Vibrio cholerae. Curr. Opin. Microbiol. 6: 35-42. Deng, W., Burland, V., Plunkett 3rd, G., Boutin, A., Mayhew, G.F., Liss, P., Perna, N.T., Rose, D.J., Mau, B., Zhou, S., Schwartz, D.C., Fetherston, J.D., Lindler, L.E., Brubaker, R.R., Plano, G.V., Straley, S.C., McDonough, K.A., Nilles, M.L., Matson, J.S., Blattner, F.R. and Perry, R.D. (2002). Genome sequence of Yersinia pestis KIM. J. Bacteriol. 184: 4601-4611. Doolittle, W.F. (1999). Phylogenetic classification and the universal tree. Science 284: 2124-2129. Figueroa-Bossi, N., Uzzau, S., Maloriol, D. and Bossi, L. (2001). Variable assortment of prophages provides a transferable repertoire of pathogenic determinants in Salmonella. Mol. Microbiol. 39: 260-271. Hayashi, T., Makino, K., Ohnishi, M., Kurokawa, K., Ishii, K., Yokoyama, K., Han, C.G., Ohtsubo, E., Nakayama, K., Murata, T., Tanaka, M., Tobe, T., Iida, T., Takami, H., Honda, T., Sasakawa, C., Ogasawara, N., Yasunaga, T., Kuhara, S., Shiba, T., Hattori, M. and Shinagawa, H. (2001). Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 8: 11-22. Hill, C.W., Sandt, C.H. and Vlazny, D.A. (1994). Rhs elements of Escherichia coli: a family of genetic composites each encoding a large mosaic protein. Mol. Microbiol. 12: 865-871. Hill, C.W., Feulner, G., Brody, M.S., Zhao, S., Sadosky, A.B. and Sandt, C.H. (1995). Correlation of Rhs elements with Escherichia coli population structure. Genetics 141: 15-24. Michel-Briand, Y. and Baysse, C. (2002). The pyocins of Pseudomonas aeruginosa. Biochimie 84: 499-510. Morgan, G.J., Hatfull, G.F., Casjens, S. and Hendrix, R.W. (2002). Bacteriophage Mu genome sequence: analysis and comparison with Mu-like prophages in Haemophilus, Neisseria and Deinococcus. J. Mol. Biol. 317: 337-359. Nakayama, K., Takashima, K., Ishihara, H., Shinomiya, T., Kageyama, M., Kanaya, S., Ohnishi, M., Murata, T., Mori, H. and Hayashi, T. (2000). The R-type pyocin of Pseudomonas aeruginosa is related to P2 phage, and the F-type is related to lambda phage. Mol. Microbiol. 38: 213-231. Nguyen, H.A., Tomita, T., Hirota, M., Kaneko, J., Hayashi, T. and Kamio, Y. (2001). DNA inversion in the tail fiber gene alters the host range specificity of carotovoricin Er, a phage-tail-like bacteriocin of phytopathogenic Erwinia carotovora subsp. carotovora Er. J. Bacteriol. 183: 6274-6281. Parkhill, J., Dougan, G., James, K.D., Thomson, N.R., Pickard, D., Wain, J., Churcher, C., Mungall, K.L., Bentley, S.D., Holden, M.T., Sebaihia, M., Baker, S., Basham, D., Brooks, K., Chillingworth, T., Connerton, P., Cronin, A., Davis, P., Davies, R.M., Dowd, L., White, N., Farrar, J., Feltwell, T., Hamlin, N., Haque, A., Hien, T.T., Holroyd, S., Jagels, K., Krogh, A., Larsen, T.S., Leather, S., Moule, S., O’Gaora, P., Parry, C., Quail, M., Rutherford, K., Simmonds, M., Skelton, J., Stevens, K., Whitehead, S. and Barrell, B.G. (2001). Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 413: 848-852. Porwollik, S. and McClelland, M. (2003). Lateral gene transfer in Salmonella. Microbes Infect. 5: 977-989. Rishovd, S., Holzenburg, A., Johansen, B.V. and Lindqvist, B.H. (1998). Bacteriophage P2 and P4 morphogenesis: structure and function of the connector. Virology 245: 11-17. Rohwer, F. (2003). Global phage diversity. Cell 113: 141. Rucinsky, T.E. and Cota-Robles, E.H. (1973). The intracellular organization of bacteriophage tail-like particles in cells of Chromobacterium violaceum following mitomycin C treatment. J. Ultrastruct. Res. 43: 260-269. Rucinsky, T.E., Gregory, J.P. and Cota-Robles, E.H. (1972). Organization of bacteriophage tail-like particles in cells of Chromobacterium violaceum. J. Bacteriol. 110: 754-757. Salanoubat, M., Genin, S., Artiguenave, F., Gouzy, J., Mangenot, S., Arlat, M., Billault, A., Brottier, P., Camus, J.C., Cattolico, L., Chandler, M., Choisne, N., Claudel-Renard, C., Cunnac, S., Demange, N., Gaspin, C., Lavie, M., Moisan, A., Robert, C., Saurin, W., Schiex, T., Siguier, P., Thebault, P., Whalen, M., Wincker, P., Levy, M., Weissenbach, J. and Boucher, C.A. (2002). Genome sequence of the plant pathogen Ralstonia solanacearum. Nature 415: 497-502. Vasconcelos, A.T.R., Almeida, D.F., Almeida, F.C., Almeida, L.G.P., Almeida, R., Alves-Gomes, J.A., Andrade, E.M., Antônio, R.V., Araripe, J., Araújo, M.F.F., Astolfi-Filho, S., Azevedo, V., Baptista, A.J., Bataus, L.A.M., Batista, J.S., Beló, A., van den Berg, C., Blamey, J., Bogo, M., Bonatto, S., Bordignon, J., Brito, C.A., Brocchi, M., Burity, H.A., Camargo, A.A., Cardoso, D.D.P., Carneiro, N.P., Carraro, D.M., Carvalho, C.M.B., Cascardo, J.C.M., Cavada, B.S., Chueire, L.M.O., Creczynski-Pasa, T.B., Duran, N., Fagundes, N., Falcão, C.L., Fantinatti, F., Farias, I.P., Felipe, M.S.S., Ferrari, L.P., Ferro, J.A., Ferro, M.I.T., Franco, G.R., Freitas, N.S.A., Furlan, L.R., Gazzinelli, R.T., Gomes, E.A., Gonçalves, P.R., Grangeiro, T.B., Grattapaglia, D., Grisard, E.C., Guimarães, C.T., Hanna, E.S., Hungria, M., Jardim, S.N., Laurino, J., Leoi, L.C.T., Lima, L.F.A., Loureiro, M.F., Lyra, M.C.C.P., Macedo, M., Madeira, H.M.F., Manfio, G.P., Maranhão, A.Q., Martins, W.S., di Mauro, S.M.Z., Medeiros, S.R.B., Meissner, R.V., Moreira, M.A.M., Nascimento, F.F., Nicolas, M.F., Oliveira, J.G., Oliveira, S.C., Paixão, RFC, Parente, J.Á., Pedrosa, F.O., Pena, S.D.J., Pereira, J.O., Pereira, M., Pinto, L.S.R.C., Pinto, L.S., Porto, J.I.R., Potrich, D.P., Ramalho-Neto, C.E., Reis, A.M.M., Rigo, L.U., Rondinelli, E., Santos, E.B.P., Santos, F.R., Schneider, M.P.C., Seuanez, H.N., Silva, A.M.R., Silva, A.L.C., Silva, D.W., Silva, R., Simões, I.D.C., Simon, D., Soares, C.M.A., Soares, R.B.A., Souza, E.M., Souza, K.R.L., Souza, R.C., Steffens, M.B.R., Steindel, M., Teixeira, S.R., Urmenyi, T., Vettore, A., Wassem, R., Zaha, A. and Simpson, A.J.G. (2003). Complete genome sequence of Chromobacterium violaceum reveals remarkable and exploitable bacterial adaptability. Proc. Nat. Acad. Sci. USA 100: 11660-11665. Wagner, P.L. and Waldor, M.K. (2002). Bacteriophage control of bacterial virulence. Infect. Immun. 70: 3985-3993. Wang, Y.D., Zhao, S. and Hill, C.W. (1998). Rhs elements comprise three subfamilies which diverged prior to acquisition by Escherichia coli. J. Bacteriol. 180: 4102-4110. Zhao, S., Sandt, C.H., Feulner, G., Vlazny, D.A., Gray, J.A. and Hill, C.W. (1993). Rhs elements of Escherichia coli K-12: complex composites of shared and unique components that have different evolutionary histories. J. Bacteriol. 175: 2799-2808. |
|