![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
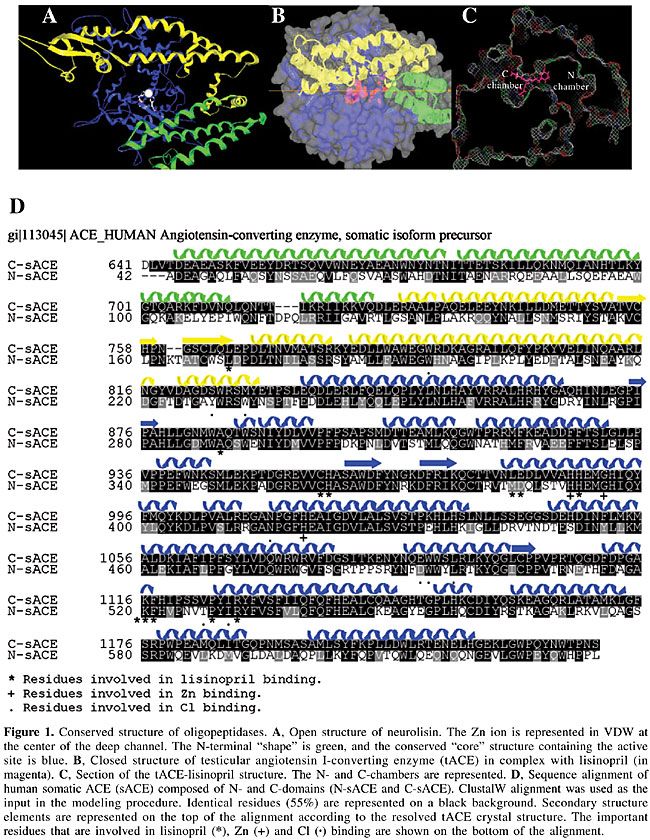
ABSTRACT. Angiotensin I-converting enzyme (ACE) is a dipeptidyl-carboxypeptidase expressed in endothelial, epithelial and neuroepithelial cells. It is composed of two domains, known as N- and C-domains, and it is primarily involved in blood pressure regulation. Although the physiological functions of ACE are not limited to its cardiovascular role, it has been an attractive target for drug design due to its critical role in cardiovascular and renal disease. We examined natural structures based on bradykinin-potentiating peptides (BPPs) extracted from Bothrops jararaca venom for ACE inhibition. Modeling, docking and molecular dynamics were used to study the conserved residues in the S2’, S1’ and S1 positions that allow enzyme-substrate/inhibitor contacts. These positions are conserved in other oligopeptidases, and they form tight and non-specific contacts with lisinopril, enalapril and BPP9a inhibitors. The only specific inhibitor for human somatic ACE (sACE) was BPP9a, which is instable in the N-sACE-BPP9a complex due to repulsive electrostatic interactions between Arg P4-Arg 412 residues. Specificity for the C-terminal domain in human sACE inhibition was confirmed by electrostatic interaction with the Asp 1008 residue. Peptide-like BPP structures, naturally developed by snakes across millions of years of evolution, appear to be good candidates for the development of domain-selec tive ACE inhibitors with high stability and improved pharmacological profiles. Key words: Bradykinin-potentiating peptides, Hypertension, Modeling, Angiotensin I-converting enzyme, Molecular dynamics INTRODUCTION During evolution, poisonous snakes became specialized in the production of a number of toxins that disorganize the physiological levels of hormones by disturbing the activity of critical enzymes, receptors, or ion channels, thus disarranging the entire cardiovascular or nervous systems of their victims. Due to their high degree of target specificity, snake venom toxins have been increasingly used as pharmacological tools and as prototypes for drug development (Camargo and Hayashi, 2005). In 1949, Rocha e Silva discovered that bradykinin, a hypotensive peptide, is produced when the venom of Bothrops jararaca is injected into the blood circulation of mammals (Rocha e Silva et al., 1949). This important bioactive peptide is involved in the control of blood pressure and in many other physiological and pathological processes. Later, in 1965, his student and collaborator, Sergio Ferreira, discovered that this venom not only generates bradykinin but it also strongly enhances its hypotensive effects through the formation of bradykinin-potentiating peptides (BPPs) (Ferreira et al., 1970). BPPs as leading structures for hypertensive treatment The synergy of BPPs causes a vascular shock in the snake’s prey, which are usually small mammals. The pharmacological and molecular features of this reaction spotlighted angiotensin I-converting enzyme (ACE) as the key enzyme for the treatment of human hypertension (Ng and Vane, 1970). The BPPs were also essential for the development of the first commercial ACE inhibitor (ACEI), captopril, for the treatment of human hypertension (Ondetti and Cushman, 1981). ACE is expressed in endothelial, epithelial and neuroepithelial cells as a 150- to 180-kDa protein (somatic) and as a smaller 90- to 110-kDa (testicular) isoform in male germinal cells. Somatic ACE (sACE) is composed of two domains, known as N- and C-domains (N-sACE and C-sACE), and testicular ACE (tACE) contains a single domain that shows high sequence identity to the C-terminal domain of sACE. Both domains have dipeptidyl-carboxypeptidase activity (Liu et al., 2001), possessing the characteristic gluzincin HExxH(x)23E motif (Wei et al., 1991). The HExxH is a canonical Zn-binding motif found in metalloproteases. Despite the high degree of sequence identity between the N- and C-domains, they differ in substrate/inhibitor specificity (Cotton et al., 2002; Hayashi et al., 2003). The N-domain is specific for the degradation of the AcSDKP tetrapeptide that controls hematopoietic stem cell proliferation and differentiation (Rousseau et al., 1995), whereas the C-domain is primarily involved in blood pressure regulation through the degradation of Ang I (a potent vasoconstrictor; Jaspard et al., 1993) and the inactivation of vasodilator peptide bradykinin (Villard and Soubrier, 1996). Although the physiological functions of ACE are not limited to its cardiovascular role, it has been an attractive target for drug design due to its critical role in cardiovascular and renal disease (Ng and Vane, 1970). ACEIs have been used for the treatment of heart diseases and hypertension for many years (reviewed in Riordan, 2003). Our interest was focused on the specificity of BPPs in the inhibition of both N- and C-terminal active sites of human sACE and on the structurally important residues contacting the substrate/inhibitors. Using modeling and docking procedures, we constructed the N- and C-sACE domains “in silico” to study the important inhibitor-contacting residues in C-sACE. Surprisingly, the data obtained for the active site demonstrate that S2’ and S1’ contacting residues are conserved not only in N- and C-sACE, but also in other important oligopeptidases described in the literature. MATERIAL AND METHODS Protein structures used in experiments We used the X-ray structures of C-domains of human C-sACE (Natesh et al., 2003, 2004), their human homologous enzyme ACE2 (Towler et al., 2004), Drosophila ACE (AnCE) (Kim et al., 2003), neurolysin (Nls) (Brown et al., 2001), and human thimed oligopeptidase (TOP) (Ray et al., 2004) deposited in the PDB databank. The N-sACE was modeled as described in Fernandez et al. (2003). BPP inhibitors (Ianzer et al., 2004) were docked in the structures that were obtained, using the Autodock 3.0 package (Morris et al., 1998). Alignments Modeling of the N-terminal domain of human sACE has already been published (Fernandez et al., 2003). Sequences of the N- and C-domains of the human sACE (P12821) and human tACE (Natesh et al., 2003) were aligned using ClustalW (Thompson et al., 1997). The alignment was used in the modeling procedure, taking the tACE structure as template (pdb 1o86) (Natesh et al., 2003). Modeling human ACE structure The structure of the tACE (pdb 1o86; Nathesh et al., 2003) has been used to predict the structure of the human N-sACE. We have taken advantage of the 55% sequence identity between these sequences to map the N-sACE sequence in the three-dimensional structure described for tACE (Nathesh et al., 2003). The atomic coordinates of the 1o86 pdb structure were used as a template in comparative modeling by satisfaction of spatial restraints (Sali and Blundell, 1993) implemented in the program MODELLER 6v2 (Fiser et al., 2000). The stereochemical quality of the five best-scoring models was assessed by PROCHECK (Laskowski et al., 1993) at a 2.0-Å resolution. The model that was obtained was subjected to relaxation and energy minimization of the structure using the GROMACS molecular dynamics package (Lindahl et al., 2001). Additional energy minimization and equilibrating molecular dynamics simulations were carried out with the GROMACS 3.2 molecular dynamics package (Lindahl et al., 2001) on a Linux workstation in order to refine the molecular model of the N-domain structure. The initial protein model was submitted to a steepest-descent energy minimization (5000 steeps) to remove bad van der Waals contacts. In order to further relax the structure that was obtained, an unrestrained molecular dynamic (MD) was performed for 200 picoseconds (ps) in water (spc model) with Berendsen-type temperature (312 K) and pressure (1 atm) coupling, in a 32 x 32 x 32 nanometer (nm) simulation cell (Louie et al., 2002), implementing the PME method (Essman et al., 1995) in the OPLS-AA force field (Lindahl et al., 2001). The same conditions were used to “denature” MD at 400 K for 200 ps and in the subsequent “refolding” stage of MD at 312 K for 200 ps. An unrestrained multiple step conjugate-gradient minimization process was used (to 0.1 kJ mol-1 nm-1) to obtain the energy minimized structure of our protein. These models are available for non-profit use upon direct request to the authors. Analysis of the dynamics of C-sACE-BPP and N-sACE-BPP Molecular dynamics simulations were initiated under the periodic boundary conditions in isothermal-isobaric ensemble with Berendsen-type temperature (312°K) and pressure (1 atm) coupling. The ensemble, with a constant number of particles, volume and temperature (NVT), was used after the density became balanced. The initial refinement process of the model takes a total time of 600 ps in the relaxation, denaturing and refolding procedures. The entire MD simulation was 1.5 ns long. The first nanosecond was used for the equilibration of the isothermal-isobaric (NVT) system, and the last 500 ps were considered to be for the production part of the dynamics. The g_cluster script, included with the Gromacs distribution, was used for cluster analysis of the trajectories. The evaluation of the inter-residue distances through the complex in the dynamics experiments was performed using the g_mindist script. The molecular dynamics trajectories were visualized by using the VMD program (Humphrey et al., 1996), and the Gold STING package (Neshich et al., 2003) was used for evaluation of residue interactions in final structures. The cutoff distances for hydrogen bonds, salt bridges and electrostatic interactions were 3.2, 4.0, and 4.0 Å, respectively. RESULTS AND DISCUSSION Substrate binding in oligopeptidases Our study focused on structural similarities in secondary elements surrounding the active site and on conserved residues of human N-sACE and C-sACE (Fernandez et al., 2003, Natesh et al., 2003, 2004), their human homologous enzyme ACE2 (Towler et al., 2004), Drosophila ACE (AnCE) (Kim et al., 2003), neurolysin (Nls) (Brown et al., 2001), and human thimed oligopeptidase (TOP) (Ray et al., 2004). Despite low amino acid sequence identity, the structural similarity of all these enzymes is remarkable, suggesting that these enzymes evolved from a common ancestor by divergent evolution. The structural similarity of the secondary structure elements is especially high in the region surrounding the active site (Figure 1A and B) and was named conserved core.
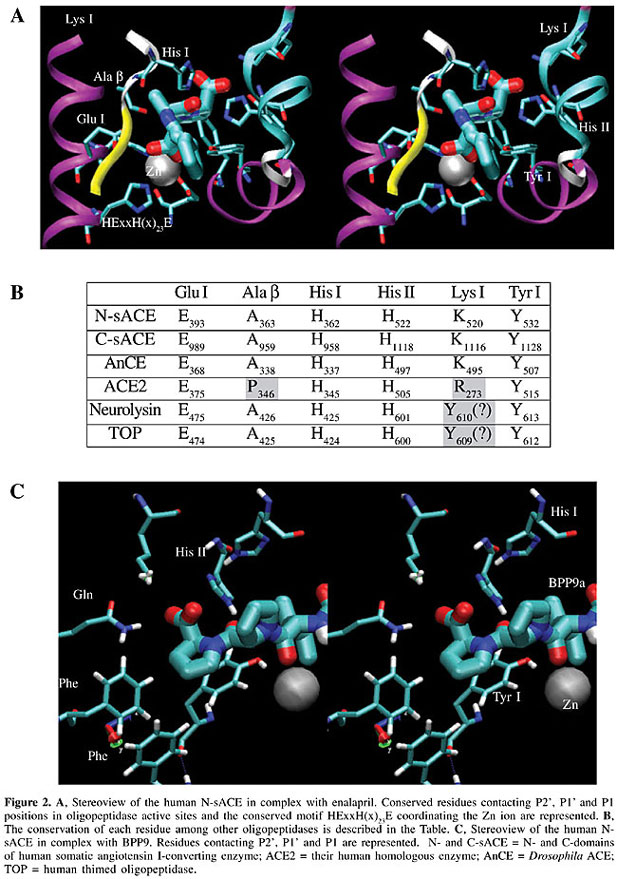
The center of the active site in all these enzymes is formed by a conserved HExxH(x)23E motif coordinating the Zn ion in the center of a deep channel (Figure 1A), and substrates or peptide inhibitors are expected to extend largely along this channel. For practical purposes, the channel is subdivided into the C-chamber, comprising the S2’ and S1’ sites, and the N-chamber, comprising the S1, S2, ..., S8 sites, contacting substrate/inhibitor sites (Figure 1C). The enzyme-substrate/inhibitor complex is formed in the open structure of the enzyme. The substrate is received by the N-terminal a-helix (Figure 1A, B, D), and placed in the bottom of the channel at the time that the enzyme is closing (Figure 1B). The substrate/inhibitor is accommodated near the active site by forming a hydrogen bond network with the residues forming the b-barrel, conserved in all the structures near the catalytic Zn ion (Figure 2A). Residues contacting the substrate/inhibitor in S2’ and S1’ positions (nomenclature in Figure 2A, B) were studied, and are conserved in different oligopeptidases (Table in Figure 2B). In the accommodation of the P2’ site (the carboxy-terminal residue) of the substrate/inhibitor, the hydrophobic contacts with Tyr I, electrostatic interactions of the carboxylic group with the Lys I residue and a hydrogen bond with the His II residue (Figure 2A, B and C) are essential. The P1’ position is coordinated by the hydrogen bonds with the His I residue and Ala b, and the carbonyl group of the scissile bond in the substrate, or the sulfhydryl, carboxyl or phosphoric groups in the inhibitors, coordinating the catalytic Zn ion (see Figure 2C for BPP9a contacts).
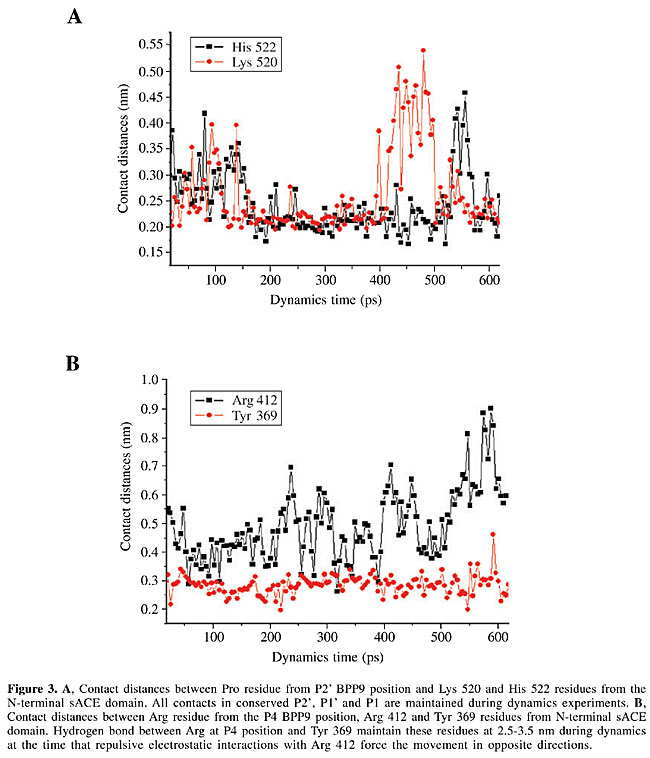
All these contacts are maintained in our dynamics experiments as demonstrated in the data on distances between the residues in the human N- and C-sACE-inhibitor complexes (Figure 3).
Specific inhibitors for the C-terminal domain of human sACE Until now, commercial inhibitors of human sACE were developed as chelating agents of reactive Zn ions. They are variations of small peptide-like structures, with a proline residue at position P2’, and are primarily directed to bind Zn ion through sulfhydryl, carboxyl or phosphoric groups. These small structures focus their binding complementarities on S2’, S1’ and S1 sites (Natesh et al., 2003, 2004). In their calorimetric studies of ACEI complexes with lisinopril, enalapril and captopril, Andújar-Sánchez et al. (2004) concluded that the contribution of the enthalpic interactions to the energetics of binding is overcome by the hydrophobic contribution. This evidence, together with our data, led us to conclude that in the accommodation of the S2’, S1’ and S1 residues of substrate/inhibitor in the final position, the hydrogen bond network and hydrophobic interactions are more important than the electrostatic interactions. These positions are conserved in different oligopeptidases (Figure 2A and B), and, for exclusion, desired enzyme selectivity is mainly determined by the S2, S3, S4, etc., interaction sites, placed at the N-terminal chamber of the enzyme (Figure 1C). The development of domain-selective ACEIs with better stability and improved pharmacological profile is facilitated when it is based on BPP structures, naturally developed by snakes during millions of years of evolution (Cotton et al., 2002; Hayashi et al., 2003). Until now, more than 15 different BPP structures have been extracted from B. jararaca venom. They are rich in proline small peptides from 5 to 13 residues, with a well-defined carboxy-terminal Ile-Pro-Pro motif and pyroglutamate at the amino-terminal (for reviews, see Ianzer et al., 2004 and Camargo and Hayashi, 2005). Among these molecules, inhibitors were found to be able to selectively inhibit N- or C-sACE (Cotton et al., 2002) and display different hypotensive effects in rats (Hayashi et al., 2003). As an example, we studied the BPP9a (Ianzer et al., 2004), a C-sACE selective inhibitor with the sequence <EWPRPQIPP. In N- and C-sACE, all the contacts in the S2’ and S1’ conserved positions were maintained in our dynamics experiments (Figure 3A), but the Arg repulsive contact in position P4 with Arg 412 of the N-sACE was reconfigured during dynamics due to the repulsive electrostatic interaction (Figure 3B). This contact was maintained in the C-sACE due to the substitution of Arg 412-Asp 1008 in the S4 contact position of N-sACE-C-sACE, respectively. The Arg-Asp substitution in the S4 position of sACE domains is, in our opinion, an important factor that allows for more specific C-sACE inhibition of human sACE by the BPP9a peptide (Ianzer et al., 2004). ACKNOWLEDGMENTS Research supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) through the Center for Applied Toxinology (CAT/CEPID) and a Post-doctoral grant 03/00785-6. Special acknowledgments go to Neusa Lima and Patricia E.F. Moraes for secretarial work. REFERENCES Andújar-Sánchez, M., Cámara-Artigas, A. and Jará-Pérez, V. (2004). A calorimetric study of the binding of lisinopril, enalaprilat and captopril to angiotensin-converting enzyme. Biophys. Chem. 111: 183-189. Brown, C.K., Madauss, K., Lian, W., Beck, M.R., Tolbert, W.D. and Rodgers, D.W. (2001). Structure of neurolysin reveals a deep channel that limits substrate access. Proc. Natl. Acad. Sci. USA 98: 3127-3132. Camargo, A.C.M. and Hayashi, M.A.F. (2005). The bradykinin-potentiating peptides from the venom gland and brain of Bothrops jararaca contain highly site-specific inhibitors of the somatic angiotensin-converting enzyme. Toxicon (in press). Cotton, J., Hayashi, M.A., Cuniasse, P., Vazeux, G., Ianzer, D., Camargo, A.C.M. and Dive, V. (2002). Selective inhibition of the C-domain of angiotensin I converting enzyme by bradykinin potentiating peptides. Biochemistry 41: 6065-6071. Essman, U., Perela, L., Berkowitz, M.L., Darden, T., Lee, H. and Pedersen, L.G. (1995). A smooth particle mesh Ewald method. J. Chem. Phys. 103: 8577-8592. Fernandez, J.H., Hayashi, M.A., Camargo, A.C.M. and Neshich, G. (2003). Structural basis of the lisinopril-binding specificity in N- and C-domains of human somatic ACE. Biochem. Biophys. Res. Commun. 22: 219-226. Ferreira, S.H., Bartelt, D.C. and Greene, L.J. (1970). Isolation of bradykinin-potentiating peptides from Bothrops jararaca venom. Biochemistry 9: 2583-2593. Fiser, A., Do, R.K. and Sali, A. (2000). Modeling of loops in protein structures. Protein Sci. 9: 1753-1773. Hayashi, M.A., Murbach, A.F., Ianzer, D., Portaro, F.C., Prezoto, B.C., Fernandez, B.L., Silveira, P.F., Silva, C.A., Pires, R.S., Britto, L.R., Dive, V. and Camargo, A.C.M. (2003). The C-type natriuretic peptide precursor of snake brain contains highly specific inhibitors of the angiotensin-converting enzyme. J. Neurochem. 85: 969-977. Humphrey, W., Dalke, A. and Schulten, K. (1996). “VMD - Visual Molecular Dynamics”. J. Mol. Graph. 14: 33-38. Ianzer, D., Konno, K., Marques-Porto, R., Vieira Portaro, F.C., Stocklin, R., Martins de Camargo, A.C. and Pimenta, D.C. (2004). Identification of five new bradykinin potentiating peptides (BPPs) from Bothrops jararaca crude venom by using electrospray ionization tandem mass spectrometry after a two-step liquid chromatography. Peptides 25: 1085-1092. Jaspard, E., Wei, L. and Alhenc-Gelas, F. (1993). Differences in the properties and enzymatic specificities of the two active sites of angiotensin I-converting enzyme (kininase II). Studies with bradykinin and other natural peptides. J. Biol. Chem. 268: 9496-9503. Kim, H.M., Shin, D.R., Yoo, O.J., Lee, H. and Lee, J.O. (2003). Crystal structure of Drosophila angiotensin I-converting enzyme bound to captopril and lisinopril. FEBS Lett. 538: 65-70. Laskowski, R.A., MacArthur, M.W., Moss, D.S. and Thornton, J.M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26: 283-291. Lindahl, E., Hess, B. and van der Spoel, D. (2001). GROMACS 3.0: A Package for molecular simulation and trajectory analysis. J. Mol. Mod. 7: 306-317. Liu, X., Fernandez, M., Wouters, M.A., Heyberger, S. and Husain, A. (2001). Arg(1098) is critical for the chloride dependence of human angiotensin I-converting enzyme C-domain catalytic activity. J. Biol. Chem. 276: 33518-33525. Louie, T.M., Yang, H., Karnchanaphanurach, P., Xie, X.S. and Xun, L. (2002). FAD is a preferred substrate and an inhibitor of Escherichia coli general NAD(P)H:flavin oxidoreductase. J. Biol. Chem. 277: 39450-39455. Morris, G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K. and Olson, A.J. (1998). Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem.19: 1639-1662. Natesh, R., Schwager, S.L.U., Sturrock, E.D. and Acharya, K.R. (2003). Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature 421: 551-554. Natesh, R., Schwager, S.L.U., Evans, H.R., Sturrock, E.D. and Acharya, K.R. (2004). Structural details on the binding of antihypertensive drugs captopril and enalaprilat to human testicular angiotensin I-converting enzyme. Biochemistry 43: 8718-8724. Neshich, G., Togawa, R.C., Mancini, A.L., Kuser, P.R., Yamagishi, M.E., Pappas Jr., G., Torres, W.V., Fonseca e Campos, T., Ferreira, L.L., Luna, F.M., Oliveira, A.G., Miura, R.T., Inoue, M.K., Horita, L.G., de Souza, D.F., Dominiquini, F., Alvaro, A., Lima, C.S., Ogawa, F.O., Gomes, G.B., Palandrani, J.F., dos Santos, G.F., de Freitas, E.M., Mattiuz, A.R., Costa, I.C., de Almeida, C.L., Souza, S., Baudet, C. and Higa, R.H. (2003). STING Millennium: A web-based suite of programs for comprehensive and simultaneous analysis of protein structure and sequence. Nucleic Acids Res. 31: 3386-3392. Ng, K.K. and Vane, J.R. (1970). Some properties of angiotensin converting enzyme in the lung in vivo. Nature 225: 1142-1144. Ondetti, M.A. and Cushman, D.W. (1981). Angiotensin converting enzyme inhibitors: biochemical properties and biological activities. In: Biochemical Regulation of Blood Pressure (Soffer, R.L., ed.). Wiley, New York, NY, USA, pp. 165-204. Ray, K., Hines, C.S., Coll-Rodriguez, J. and Rodgers, D.W. (2004). Crystal structure of human thimet oligopeptidase provides insight into substrate recognition, regulation, and localization. J. Biol. Chem. 279: 20480-20489. Riordan, J.F. (2003). Angiotensin-I-converting enzyme and its relatives. Genome Biol. 4: 225-229. Rocha e Silva, M., Beraldo, W.T. and Rosenfeld, G. (1949). Bradykinin, a hypotensive and a smooth muscle stimulating factor released from plasma globulin by snake venom and by trypsin. Am. J. Physiol. 156: 261-270. Rousseau, A., Michaud, A., Chauvet, M.T., Lenfant, M. and Corvol, P. (1995). The hemoregulatory peptide N-acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate of the N-terminal active site of human angiotensin-converting enzyme. J. Biol. Chem. 270: 3656-3661. Sali, A. and Blundell, T.L. (1993). Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 234: 779-815. Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F. and Higgins, D.G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25: 4876-4882. Towler, P., Staker, B., Prasad, S.G., Menon, S., Tang, J., Parsons, T., Ryan, D., Fisher, M., Williams, D., Dales, N.A., Patane, M.A. and Pantoliano, M.W. (2004). ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 279: 17996-18007. Villard, E. and Soubrier, F. (1996). Molecular biology and genetics of the angiotensin-I-converting enzyme: potential implications in cardiovascular diseases. Cardiovasc. Res. 32: 999-1007. Wei, L., Alhenc-Gelas, F., Corvol, P. and Clauser, E. (1991). The two homologous domains of human angiotensin I-converting enzyme are both catalytically active. J. Biol. Chem. 266: 9002-9008. |
|