![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
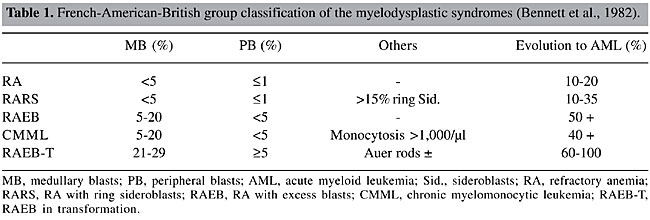
ABSTRACT. The myelodysplastic syndromes (MDS) are clonal hematopoietic diseases characterized by medullary dysplasia, cytopenias, and frequent evolution to acute myeloid leukemia. In 1982, the French-American-British (FAB) group proposed a classification for the MDS, based on morphological characteristics of peripheral blood and of the bone marrow. Later, cytogenetics proved to be a useful tool for the refinement of prognosis, through the use of the International Prognosis Score System (IPSS), as well as through evidence of clonality. Recently, the World Health Organization (WHO) proposed a new classification for the MDS, based on significant modifications of the FAB proposal, with the inclusion of chromosome analysis. A cytogenetic analysis was made of 17 patients with symptoms of MDS in the State of Pará, based on WHO recommendations, and application of the IPSS. Good metaphases were obtained for 13 patients; 12 had a normal karyotype and only one had a clonal abnormality, del(3)(p25). The genes related to neoplastic processes that have been mapped to 3p are: XPC in 3p25.1 and FANCD2 and VHL in 3p25-26. Four patients had classic symptoms of MDS; in the rest the possibility of MDS was excluded or several months of observation before diagnosis were recommended. Among those with MDS, it was not possible to apply IPSS and WHO recommendations, because fundamental data were lacking, specifically the medullary blast and ring sideroblast counts. We advocate the implementation of routine cytogenetic analyses for the study of MDS, especially in patients with moderate hematopoietic dysplasia. Key words: Myelodysplastic syndromes, Cytogenetics, Classification, del(3)(p25) INTRODUCTION The myelodysplastic syndromes (MDS) are clonal diseases of hematopoietic stem cells, initially characterized by inefficient hematopoiesis, of one or more cell lines, and peripheral cytopenias, as well as a high risk of progression to acute myeloid leukemia (AML) (Dunbar and Saunthararajah, 2000). In 1982, the French-American-British group (FAB; Bennett et al., 1982) proposed a classification for the MDS, based on morphological characteristics in the peripheral blood (PB) and the bone marrow (BM), in which they defined five distinct subtypes with different prognostic values (Table 1).
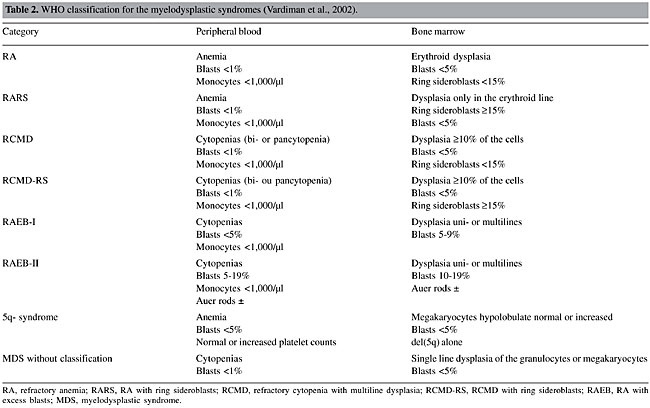
This classification was widely accepted by pathologists and clinicians and was combined with the International Prognostic Scoring System (IPSS) (Greenberg et al., 1997), which utilizes the percentage medullary blasts, the number of cytopenic lines and cytogenetic abnormalities as prognostic factors, signifying an advance in the diagnosis and treatment of patients with MDS. Nevertheless, with time, some difficulties appeared. The heterogeneity of refractory anemias, the inclusion and definition of chronic myelomonocytic leukemia (CMML) in the FAB system, the definition of prognosis of patients with prognosis of del(5q), and the fact that patients with medullary blast counts above 20% generally have a prognosis similar to one with AML, are some examples (Germing et al., 2000; Steensma and Tefferi, 2003). For these reasons, the FAB proposal was recently revised by World Health Organization (WHO) specialists, resulting in a new classification for the MDS, with more homogeneous categories (Table 2) (Bennett, 2000; Germing et al., 2000; Harris et al., 2000; Jaffe et al., 2001; Vardiman et al., 2002).
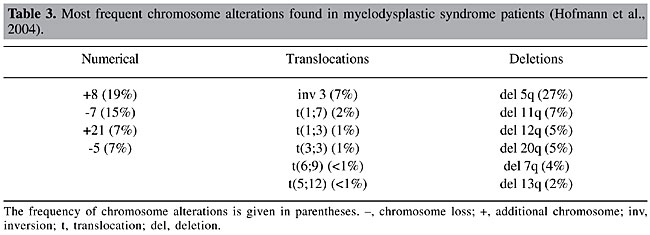
The main changes to the WHO classification refined the definitions of low-grade subtypes (refractory anemia (RA) and RA with ring sideroblasts (RARS)) as being strictly erythrocytic, and added a new category, refractory cytopenia with multiline dysplasia (RCMD). The lower limit of percentage blasts for the diagnosis of AML, in PB or BM, was reduced from 30 to 20%, which resulted in the elimination of subtype RA with excess blasts in transformation (RAEB-T) from the WHO classification. Other changes included the recognition of new subtypes: two subtypes of RAEB; RAEB-I, with 5-9% medullary blasts and RAEB-II with 10-19%; MDS without classification, and the specific genetic subtype, 5q- syndrome (Vardiman et al., 2002). This new classification scheme is not universally accepted; while it is criticized by some researchers (Greenberg et al., 2000; Nosslinger et al., 2001), it is supported by others (Germing et al., 2000; Bennett et al., 2002; Dunkley et al., 2002; Howe et al., 2003; Strupp et al., 2003; Giagounidis et al., 2004). Different from the FAB proposal, the WHO classification includes a genetic subcategory specific for MDS. Consequently, cytogenetic information is indispensable, and the unavailability on short notice of these data is an obstacle for this classification (Arber, 2001). The most common cytogenetic abnormalities in the MDS are chromosome 5, 7, 11, 12, and 20 deletions, and/or chromosome 8 trisomy (Table 3). The frequency of chromosome abnormalities in primary MDS is 30-50%, while it is 80% in secondary MDS. The latter generally has complex karyotypes (Fenaux et al., 1996; Mecucci and La Starza, 1999).
The MDS are known to have recurrent chromosome deletions. This loss of genetic material raises the hypothesis that tumor suppression genes could be involved in the pathogenic process (Hofmann et al., 2004). Table 4 lists abnormalities that are frequently found in MDS, and also lists the genes that are possibly involved in pathogenesis.
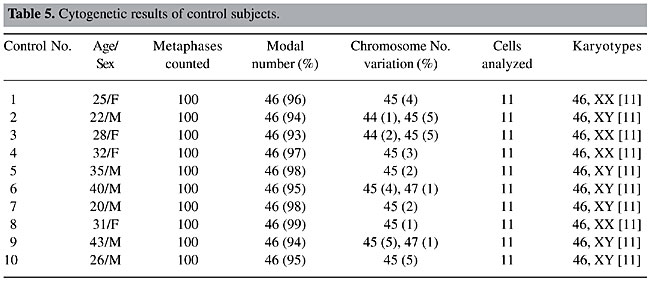
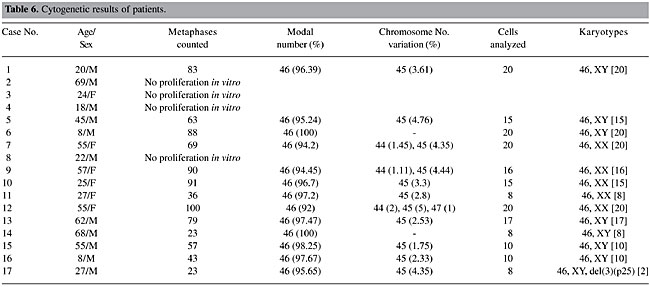
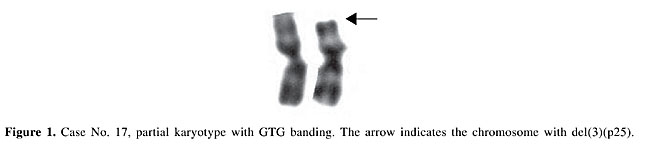
Our objective was the cytogenetic characterization of patients with signs and symptoms of MDS in the State of Pará, for confirmatory diagnosis of the clinical-morphological diagnosis, based on WHO criteria. Our specific objectives were to i) describe the recurrent break points and associate them with tumor suppressor genes, DNA repair genes, oncogenes, and other genes involved in these malignant processes; ii) relate the alterations that are observed to the evolution of the neoplasia, to determine their effect on diagnosis and prognosis, and iii) reclassify the patients that were referred based on WHO criteria and define prognosis groups based on IPSS, to develop a basis for the choice of therapy. MATERIAL AND METHODS BM and PB samples Samples of BM of 17 patients suspected or diagnosed as having MDS without previous cytotoxic therapy and samples of PB of 10 control subjects were analyzed. All the study subjects were seen from February 4, 2002 to January 21, 2003. The patients, or their legal representatives, were informed about the research and they all signed a permission form, allowing us to investigate tissue samples that would already normally be collected during routine exams, so that there was no additional discomfort for these volunteers. Methodology The BM samples of patients were cultivated in MARROWMAXTM Bone Marrow Medium (Gibco). Three cultures were made for each BM sample: direct, 24 h and 48 h. Lymphocytes from PB of control subjects, prepared from 0.5 ml heparinized blood obtained by venipuncture, were cultivated for 72 h in Ham’s F10 medium (Gibco) supplemented with 20% fetal bovine serum and antibiotics. The cells were stimulated with 2% PHA (Gibco). A 0.1-ml aliquot of colchicine (0.0016%) was added to all cell cultures, 2 h before each harvest. Posteriorly, these cells were centrifuged at 1000 rpm for 10 min, the supernatant was removed and a hypotonic KCl treatment (0.075 M) was initiated at 37°C for 20 min. The cells were then centrifuged and fixed three times with methanol/acetic acid (3:1). Cell suspensions were placed on histological slides. After drying at room temperature, the slides were stained for conventional analysis with 4% Giemsa solution, pH 6.8, for 10 min and washed with distilled water. The GTG banding technique (Scheres, 1972) was used, with some modifications. Dried slides were artificially aged overnight in a drying oven at 60°C or for 1 h at 90°C. They were then treated with 0.01% trypsin solution, and diluted in phosphate buffer, pH 6.8, for 3-5 min. The trypsin activity was halted by bathing the slides in ice-cold distilled water. After drying at room temperature, the slides were stained with 4% Giemsa solution, pH 6.8, for 10 min. The identification and classification of the chromosomes were made based on recommendations of the International System for Cytogenetics Nomenclature (ISCN, 1995), as were the criteria for the determination of cytogenetic clones. The cytogenetic data of each patient were combined with the results of their respective histopathological (BM biopsy), myelogram and hemogram, along with the clinical evaluation, for posterior analysis. Statistical analysis The data were subjected to statistical analysis, using the chi-square test and the Friedman test to compare the variation of chromosome number in the patients in relation to the respective controls. RESULTS AND DISCUSSION Cytogenetic analysis was performed in all control samples but only in 13 of the 17 patients’ samples. In the remainder we were not able to obtain cellular proliferation in vitro. The modal chromosome number was 46 in all cases (Tables 5 and 6) and no statistical differences were observed between group of patients and their respective controls (P > 0.05). Even with chromosome number variation ranging between 44 and 47 chromosomes (accept values), no numeric abnormalities were found in all cells analyzed and only one clonal abnormality was found in a patient sample, del(3)(p25) (Figure 1 and Table 6).
Six of the 10 control subjects and 11 of the 17 patients were males, giving a M/F proportion of 1.5 and 1.8, respectively. While the control group varied from 20-43 years old (mean 30.2) the patient group varied from 8-69 years old (mean 37.9). At the time the samples were sent for cytogenetic analysis, only three patients were over 60, four were between 50 and 59, and most of the patients (N = 10) were less than 50 years old. Nevertheless, MDS are considered geriatric diseases (Aul et al., 1995), with more than 80% of the patients being 60 years and older, and only 8-10% with less than 50 years at diagnosis. Based on these age data, it appears that most of our patients did not actually have MDS. Patient No. 9, for example, had hematological data typical of a myeloproliferative disease (neutrophils: 17,025/µl; monocytes: 1,362/µl; platelets: 1,544,000/µl), incompatible with what is expected for a patient with MDS. Patient No. 10 had alterations in the BM that were suspected to be idiopathic thrombocytopenic purpura (IPT). IPT and MDS appear to be completely distinct diseases; IPT is a typical autoimmune disease, while MDS is a clonal neoplastic disease; however, they may initially have indistinguishable symptoms. Myelodysplastic syndromes can initially present an isolated thrombocytopenia (Menke et al., 1992) that is indistinguishable from IPT on routine exams, which include full counts of the blood cells and examination of PB smears. For this reason, BM aspirates and biopsies, which are not routinely requested for the diagnosis of IPT, could be appropriate to exclude MDS in elderly patients (George et al., 1996). However, patient No. 10 was 25 years old, and had a normal karyotype, which precludes a neoplastic disease; therefore, the hypothesis of MDS can be excluded. Not all the conditions that have pathological characteristics similar to the MDS are clonal neoplastic diseases. Even though many proposals have been made (Culligan and Jacobs, 1992; Ost and Reizenstein, 1992; Tricot, 1992; George et al., 1996; Ramos et al., 1999; Gardais, 2000), the minimum criteria for the diagnosis of MDS are not clear. Some pathologists are uncomfortable with making this diagnosis, in the absence of dysplasia in at least two cell lines, since erythroid dysplasia has many potential etiologies. However, when non-clonal conditions are eliminated, the diagnosis still cannot be assured. The distinction between MDS and similar clonal hematopoietic diseases can also be a serious challenge, since the divisions between some chronic myeloid diseases are not clear, and sometimes they can superimpose (Neuwirtova et al., 1996; Bain, 1999). Bain (1996) and Ramos et al. (1999), based on their findings of moderate dysplastic hematopoiesis in a large proportion of normal individuals, argued in favor of a higher threshold for the morphologic diagnosis of the MDS whenever a cytogenetic exam for clones and the associated abnormalities is not available. For this reason, we followed the recommendations of Greenberg et al. (1998), members of the NCCN (National Comprehensive Cancer Network), that “when classic characteristics are lacking, the patient needs to be examined during several months in order to diagnose MDS”. Consequently, the patients that presented light to moderate degrees of dysplasia and normal karyotypes (Nos. 1, 7, 11, and 14) were excluded from further analysis. In about 90% of the cases, the patients with MDS had a normocellular or hypercellular BM. However, about 10% of the patients with MDS present medullary hypoplasia at diagnosis (Nand and Godwin, 1988; Rosati et al., 1996), indicating a cellularity of less than 30%, in general. Another characteristic that indicates that our sample is not typical is that 9 of the 17 patients (Nos. 1, 3, 4, 5, 8, 11, 15, 16, and 17) presented hypocellularity or medullary aplasia. The mean age of these patients was 27.3 years, compared with 62.1 years in the group studied by Goyal et al. (1999), composed of patients with hypoblastic MDS. Among these, the BM samples from patients 3, 4, 8 and, especially patient No. 2, which presented a hypercellular medulla, did not proliferate in vitro. This failure could be attributed to the myelofibrosis observed in patient 2, the medullary aplasia observed in patients 3 and 4, and the accentuated hypoplasia in patient No. 8. Patients 5 and 15 presented medullary hypoplasias, with no dysplasia and a normal karyotype. Therefore, the NCCN recommendation was also followed for these cases. Patient No. 16 is an eight-year-old child. For this reason, and because the child had medullary hypoplasia, the hypothesis of Fanconi anemia (FA) should be discarded. The cultivated lymphocytes of FA patients have a high prevalence of chromosome breaks, which are amplified by treatment with diepoxybutane or mitomicin C. Even though the karyotypic analysis of BM cells of patient 16 was normal, if there is suspicion of FA, a test for chromosome breaks induced by diepoxybutane or mitomicin C should be made, and not only conventional CTG banding analysis. Patient No. 6 is also an eight-year-old child; however, the hypothesis of FA can be discarded, as this patient had a hypercellular BM. Based on the available data, we consider only patients 2, 12, 13, and 17, with 69, 55, 62, and 27 years old, respectively, to have MDS. Patients 2, 12 and 13 were so-classified based on strong BM dysplasia, and patient 17, even with medullary hypoplasia and moderate erythrocytic dysplasia, based on clonal cytogenetic abnormality, del(3)(p25). Cytogenetic abnormalities that result in deletion 3p are common in solid tumors, which indicates the presence of a tumor suppression gene in this chromosome arm. Johansson et al. (1997) indicated that in hematological neoplasias, including the MDS, the breakpoints in chromosome 3 are more distal than those found in solid tumors, suggesting that different tumor suppression genes are involved in these processes. There are three genes related to neoplastic processes mapped to region del(3)(p25). i) XPC, located at 3p25.1, codes for a nuclear protein involved in the premature recognition of DNA damage in the chromatin, and which affects predisposition to xeroderma pigmentosum (Stary and Sarasin, 2001). ii) FANCD2, located at 3p25-26, near the XPC gene, codes for a nuclear protein that is part of the D complement of FA (Huret, 2002). iii) VHL, located at 3p25-26, is a multifunctional tumor suppressor, which among other functions forces the cells out of the cell cycle into quiescence (Richard, 2002). These genes are contributions of this work for further molecular studies, which are needed to demonstrate its possible biological roles in MDS pathogenesis. Six months after cytogenetic analysis of the BM of patient No. 17, there was a recommendation for an allogenic transplant, due to marked pancytopenia in the PB and intense medullary aplasia, which impeded a new karyotyping. This fact corroborates the hypothesis that del(3)(p25) is an indicator of a bad prognosis. The FAB cooperative group (Bennett et al., 1982) proposed a classification for MDS, based on morphological characteristics of the PB cells and of the BM cells, which defined five subtypes with significant differences for prognosis. Nevertheless, it was observed that even those patients with marked dysplastic characteristics (Nos. 2, 12 and 13) were diagnosed generically as having MDS, and the FAB subtypes were not distinguished. Part of this inability to classify the patients is due to failures in laboratory procedures. Except for case No. 2, the percentage blasts in the BM was not informed. This information is of fundamental importance for classification schemes (both for FAB and WHO) and to determine a prognosis for these patients, through the use of the IPSS. Another important observation is that in none of the cases was the percentage ring sideroblasts informed. This information defines the specific MDS subtypes, RARS (in the FAB and WHO classifications) and RCMD-RS (in the WHO classification). This study had as an objective, given cytogenetic data, to reclassify patients based on the WHO proposal and the IPSS. Nevertheless, this procedure would be arbitrary without information on the percentage medullary blasts and ring sideroblasts. Though its prognostic value has already been proven, the IPSS is more efficient when it is combined with a classification scheme (Greenberg et al., 1997). In fact, some studies have concluded that the IPSS is limited in its ability to make a prognosis in patients with high survival rates, as well as in cases where only the erythrocytic cell line is involved (Balduini et al., 1997; Greenberg et al., 1997; Matsuda et al., 1999). Also, many other biological characteristics that can be used for prognosis are currently known. For example, the presence of abnormal location of immature precursors observed in the BM biopsy of patient No. 13 (Bellamy et al., 2001; Verburgh et al., 2003), the methylated state of the genes CDKN2B and DAPK (Tien et al., 2001; Voso et al., 2004), the length of the telomere (Ohyashiki et al., 1999), the degree of medullary apoptosis (Shimazaki et al., 2000), the mutation state of the genes, such as RAS, FMS and TP53 (Paquette et al., 1993; Padua et al., 1998), and the degree of expression of gene WT1 (Cilloni et al., 2003) can have diagnostic value. The comparison of cytogenetic data with clinical and hematological information in patients suspected of having MDS, who were evaluated by Pará State health professionals, allows us to conclude that these patients are likely being diagnosed and treated as if they had leukemia. It is imperative that a cytogenetic analysis be made routinely of patients suspected to have MDS. ACKNOWLEDGMENTS Research supported by FINEP CT-INFRA/FADESP (No. 1017-01) and CAPES. REFERENCES Arber, D.A. (2001). Realistic pathologic classification of acute myeloid leukemias. Am. J. Clin. Pathol. 115: 552-560. Aul, C., Gattermann, N. and Schneider, W. (1995). Epidemiological and etiological aspects of myelodysplastic syndromes. Leuk. Lymphoma 16: 247-262. Bain, B.J. (1996). The bone marrow aspirate of healthy subjects. Br. J. Haematol. 94: 206-209. Bain, B.J. (1999). The relationship between the myelodysplastic syndromes and the myeloproliferative disorders. Leuk. Lymphoma 34: 443-449. Balduini, C.L., Guarnone, R., Pecci, A., Centenara, E. and Ascari, E. (1997). International prognostic scoring system and other prognostic systems for myelodysplastic syndromes. Blood 90: 4232-4234. Bellamy, W.T., Richter, L., Sirjani, D., Roxas, C., Glinsmann-Gibson, B., Frutiger, Y., Grogan, T.M. and List, A.F. (2001). Vascular endothelial cell growth factor is an autocrine promoter of abnormal localized immature myeloid precursors and leukemia progenitor formation in myelodysplastic syndromes. Blood 97: 1427-1434. Bennett, J.M. (2000). World Health Organization classification of the acute leukemias and myelodysplastic syndrome. Int. J. Hematol. 72: 131-133. Bennett, J.M., Catovsky, D., Daniel, M.T., Flandrin, G., Galton, D.A., Gralnick, H.R. and Sultan, C. (1982). Proposals for the classification of the myelodysplastic syndromes. Br. J. Haematol. 51: 189-199. Bennett, J.M., Brunning, R.D. and Vardiman, J.W. (2002). Myelodysplastic syndromes: from French-American-British to World Health Organization: a commentary. Blood 99: 3074-3075. Cilloni, D., Gottardi, E., Messa, F., Fava, M., Scaravaglio, P., Bertini, M., Girotto, M., Marinone, C., Ferrero, D., Gallamini, A., Levis, A. and Saglio, G. (2003). Significant correlation between the degree of WT1 expression and the International Prognostic Scoring System Score in patients with myelodysplastic syndromes. J. Clin. Oncol. 21: 1988-1995. Culligan, D.J. and Jacobs, A. (1992). Minimal diagnostic criteria for the myelodysplastic syndrome. Leuk. Res. 16: 4-5. Dunbar, C. and Saunthararajah, Y. (2000). Myelodysplastic syndromes. In: Bone Marrow Failure Syndromes (Young, N.S., ed.). W.B. Saunders Co., Philadelphia, PA, USA, pp. 69-98. Dunkley, S.M., Manoharan, A. and Kwan, Y.L. (2002). Myelodysplastic syndromes: prognostic significance of multilineage dysplasia in patients with refractory anemia or refractory anemia with ringed sideroblasts. Blood 99: 3870-3871 (author reply 3871). Fenaux, P., Morel, P. and Lai, J.L. (1996). Cytogenetics of myelodysplastic syndromes. Semin. Hematol. 33: 127-138. Gardais, J. (2000). Dyshaemopoiesis in adults: a practical classification for diagnosis and management. Leuk. Res. 24: 641-651. George, J.N., Woolf, S.H., Raskob, G.E., Wasser, J.S., Aledort, L.M., Ballem, P.J., Blanchette, V.S., Bussel, J.B., Cines, D.B., Kelton, J.G., Lichtin, A.E., McMillan, R., Okerbloom, J.A., Regan, D.H. and Warrier, I. (1996). Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood 88: 3-40. Germing, U., Gattermann, N., Strupp, C., Aivado, M. and Aul, C. (2000). Validation of the WHO proposals for a new classification of primary myelodysplastic syndromes: a retrospective analysis of 1600 patients. Leuk. Res. 24: 983-992. Giagounidis, A.A., Germing, U., Haase, S., Hildebrandt, B., Schlegelberger, B., Schoch, C., Wilkens, L., Heinsch, M., Willems, H., Aivado, M. and Aul, C. (2004). Clinical, morphological, cytogenetic, and prognostic features of patients with myelodysplastic syndromes and del(5q) including band q31. Leukemia 18: 113-119. Goyal, R., Qawi, H., Ali, I., Dar, S., Mundle, S., Shetty, V., Mativi, Y., Allampallam, K., Lisak, L., Loew, J., Venugopal, P., Gezer, S., Robin, E., Rifkin, S. and Raza, A. (1999). Biologic characteristics of patients with hypocellular myelodysplastic syndromes. Leuk. Res. 23: 357-364. Greenberg, P., Cox, C., LeBeau, M.M., Fenaux, P., Morel, P., Sanz, G., Sanz, M., Vallespi, T., Hamblin, T., Oscier, D., Ohyashiki, K., Toyama, K., Aul, C., Mufti, G. and Bennett, J. (1997). International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 89: 2079-2088. Greenberg, P., Bishop, M.R., Deeg, H.J., Estey, E., Erba, H., Gore, S., Nimer, S., O’Donnell, M., Tallman, M., Bennett, J., Estey, E. and Stone, R. (1998). NCCN practice guidelines for the myelodysplastic syndromes. Oncology 12: 53-80. Greenberg, P., Anderson, J., de Witte, T., Estey, E., Fenaux, P., Gupta, P., Hamblin, T., Hellstrom-Lindberg, E., List, A., Mufti, G., Neuwirtova, R., Ohyashiki, K., Oscier, D., Sanz, G., Sanz, M. and Willman, C. (2000). Problematic WHO reclassification of myelodysplastic syndromes. Members of the International MDS Study Group. J. Clin. Oncol. 18: 3447-3452. Harris, N.L., Jaffe, E.S., Diebold, J., Flandrin, G., Muller-Hermelink, H.K., Vardiman, J., Lister, T.A. and Bloomfield, C.D. (2000). The World Health Organization classification of hematological malignancies report of the Clinical Advisory Committee Meeting, Airlie House, Virginia, November 1997. Mod. Pathol. 13: 193-207. Hofmann, W.K., Lubbert, M., Hoelzer, D. and Phillip Koeffler, H. (2004). Myelodysplastic syndromes. Hematol. J. 5: 1-8. Howe, R.B., Porwit-MacDonald, A., Wanat, R., Tehranchi, R. and Hellstrom-Lindberg, E. (2003). The WHO classification of MDS does make a difference. Blood 103: 3265-3270. Huret, J.L. (2002). FANCD2 (Fanconi anemia, complementation group D2).. Atlas of Genetics and Cytogenetics in Oncology and Haematology. Available at: <http://www.infobiogen.fr/services/chromcancer/Genes/FAD.html>. Accessed March 8, 2004. ISCN (1995). An International System for Human Cytogenetic Nomenclature (Mitelman, F., ed.). S. Karger, Basel, Switzerland. Jaffe, E.S., Harris, N.L., Stein, H. and Vardiman, J.W. (2001). World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of the Haematopoietic and Lymphoid Tissues. IARC, Lyon, France. Johansson, B., Billstrom, R., Kristoffersson, U., Akerman, M., Garwicz, S., Ahlgren, T., Malm, C. and Mitelman, F. (1997). Deletion of chromosome arm 3p in hematologic malignancies. Leukemia 11: 1207-1213. Matsuda, A., Jinnai, I., Yagasaki, F., Kusumoto, S., Murohashi, I., Bessho, M., Hirashima, K., Honda, S., Minamihisamatsu, M., Fuchigami, K., Matsuo, T., Kuriyama, K. and Tomonaga, M. (1999). New system for assessing the prognosis of refractory anemia patients. Leukemia 13: 1727-1734. Mecucci, C. and La Starza, R. (1999). Cytogenetics of myelodysplastic syndromes. Forum 9: 4-13. Menke, D.M., Colon-Otero, G., Cockerill, K.J., Jenkins, R.B., Noel, P. and Pierre, R.V. (1992). Refractory thrombocytopenia. A myelodysplastic syndrome that may mimic immune thrombocytopenic purpura. Am. J. Clin. Pathol. 98: 502-510. Mhawech, P. and Saleem, A. (2001). Myelodysplastic syndrome: review of the cytogenetic and molecular data. Crit. Rev. Oncol. Hematol. 40: 229-238. Nand, S. and Godwin, J.E. (1988). Hypoplastic myelodysplastic syndrome. Cancer 62: 958-964. Neuwirtova, R., Mocikova, K., Musilova, J., Jelinek, J., Havlicek, F., Michalova, K. and Adamkov, M. (1996). Mixed myelodysplastic and myeloproliferative syndromes. Leuk. Res. 20: 717-726. Nosslinger, T., Reisner, R., Koller, E., Gruner, H., Tuchler, H., Nowotny, H., Pittermann, E. and Pfeilstocker, M. (2001). Myelodysplastic syndromes, from French-American-British to World Health Organization: comparison of classifications on 431 unselected patients from a single institution. Blood 98: 2935-2941. Ohyashiki, J.H., Iwama, H., Yahata, N., Ando, K., Hayashi, S., Shay, J.W. and Ohyashiki, K. (1999). Telomere stability is frequently impaired in high-risk groups of patients with myelodysplastic syndromes. Clin. Cancer Res. 5: 1155-1160. Ost, A. and Reizenstein, P. (1992). Minimal diagnostic criteria for the myelodysplastic syndrome. Leuk. Res. 16: 9-11. Padua, R.A., Guinn, B.A., Al-Sabah, A.I., Smith, M., Taylor, C., Pettersson, T., Ridge, S., Carter, G., White, D., Oscier, D., Chevret, S. and West, R. (1998). RAS, FMS and p53 mutations and poor clinical outcome in myelodysplasias: a 10-year follow-up. Leukemia 12: 887-892. Paquette, R.L., Landaw, E.M., Pierre, R.V., Kahan, J., Lubbert, M., Lazcano, O., Isaac, G., McCormick, F. and Koeffler, H.P. (1993). N-ras mutations are associated with poor prognosis and increased risk of leukemia in myelodysplastic syndrome. Blood 82: 590-599. Ramos, F., Fernandez-Ferrero, S., Suarez, D., Barbon, M., Rodriguez, J.A., Gil, S., Megido, M., Ciudad, J., Lopez, N., del Canizo, C. and Orfao, A. (1999). Myelodysplastic syndrome: a search for minimal diagnostic criteria. Leuk. Res. 23: 283-290. Richard, S. (2002). VHL. Atlas of Genetics and Cytogenetics in Oncology and Haematology. Available at: <http://www.infobiogen.fr/services/chromcancer/Genes/VHLID132.html>. Accessed March 8, 2004. Rosati, S., Anastasi, J. and Vardiman, J. (1996). Recurring diagnostic problems in the pathology of the myelodysplastic syndromes. Semin. Hematol. 33: 111-126. Scheres, J.M. (1972). Identification of two Robertsonian translocations with a Giemsa banding technique. Humangenetik 15: 253-256. Shimazaki, K., Ohshima, K., Suzumiya, J., Kawasaki, C. and Kikuchi, M. (2000). Evaluation of apoptosis as a prognostic factor in myelodysplastic syndromes. Br. J. Haematol. 110: 584-590. Stary, A. and Sarasin, A. (2001). XPC. Atlas of Genetics and Cytogenetics in Oncology and Haematology. <http://www.infobiogen.fr/services/chromcancer/Genes/XPCID122.html>. Consulted March 8, 2004. Steensma, D.P. and Tefferi, A. (2003). The myelodysplastic syndrome(s): a perspective and review highlighting current controversies. Leuk. Res. 27: 95-120. Strupp, C., Gattermann, N., Giagounidis, A., Aul, C., Hildebrandt, B., Haas, R. and Germing, U. (2003). Refractory anemia with excess of blasts in transformation: analysis of reclassification according to the WHO proposals. Leuk. Res. 27: 397-404. Tien, H.F., Tang, J.H., Tsay, W., Liu, M.C., Lee, F.Y., Wang, C.H., Chen, Y.C. and Shen, M.C. (2001). Methylation of the p15(INK4B) gene in myelodysplastic syndrome: it can be detected early at diagnosis or during disease progression and is highly associated with leukaemic transformation. Br. J. Haematol. 112: 148-154. Tricot, G.J. (1992). Minimal diagnostic criteria for the myelodysplastic syndrome in clinical practice. Leuk. Res. 16: 5-6. Vardiman, J.W., Harris, N.L. and Brunning, R.D. (2002). The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 100: 2292-2302. Verburgh, E., Achten, R., Maes, B., Hagemeijer, A., Boogaerts, M., De Wolf-Peeters, C. and Verhoef, G. (2003). Additional prognostic value of bone marrow histology in patients subclassified according to the International Prognostic Scoring System for myelodysplastic syndromes. J. Clin. Oncol. 21: 273-282. Voso, M.T., Scardocci, A., Guidi, F., Zini, G., Di Mario, A., Pagano, L., Hohaus, S. and Leone, G. (2004). Aberrant methylation of DAP-kinase in therapy-related acute myeloid leukemia and myelodysplastic syndromes. Blood 103: 698-700. |
|